
常染色体显性遗传性聋家系遗传学特征及外显子组测序分析*
2014-12-04 02:31王燕飞贾婧洁程静卢宇张蕾韩东一袁慧军
听力学及言语疾病杂志 2014年4期
王燕飞 贾婧洁 程静 卢宇 张蕾 韩东一 袁慧军
1 中国人民解放军总医院耳鼻咽喉头颈外科 耳鼻咽喉研究所(北京 100853)
导致耳聋的常见病因中,环境因素约占40%,遗传因素约占60%。绝大多数的遗传性聋为单基因致病,只有少数耳聋由多基因联合致病。耳聋具有明显的遗传异质性,即同一表型的耳聋可由不同的基因或突变引起,而不同表型又可由同一致病基因所引起。遗传性聋分为综合征型(30%)及非综合征型聋(70%)[1],非综合征型聋包括常染色体显性遗传性聋(DFNA)、常染色体隐性遗传性聋(DFNB)、X-连锁遗传性聋(DFNX)、Y-连锁遗传性聋(DFNY)及线粒体遗传性聋。约有20%的非综合征型聋为常染色体显性遗传[2],主要表现为语后聋,听力损失呈进行性加重。至2014年4月1日,已报道的与常染色体显性遗传非综合征型聋相关的基因座有56个,耳聋基因有30个[http://hereditaryhearingloss.org]。
近几年新一代测序技术(next generation sequencing,NGS)已经被广泛应用于遗传研究领域。全外显子组测序(whole exome sequencing,WES)是NGS技术应用的一个方面,旨在通过检测全基因组外显子及其邻近区域DNA序列,鉴定出引起蛋白功能改变的基因变异,用于常见或罕见遗传性疾病的基因鉴定工作。自2010年以来,相继报道了14个应用NGS技术新鉴定的耳聋基因,其中非综合征型聋基因11个,包括1个常染色体显性遗传耳聋基因TNC[3],8个常染色体隐性遗传耳聋基因BDP1[4]、ELMOD3[5]、OTOGL[6]、TSPEAR[7]、KARS[8]、TBC1D24[9]、GPSM2[10]、TPRN[11]及2个X-连锁遗传耳聋基因SMPX[12]和 COL4A6[13],3个引 起 Perrault综 合 征 的 基 因 HSD17B4[14]、LARS2[15]、CLPP[16],以及多个新致聋位点。
本研究对一个常染色体显性遗传性聋家系的听力学特点和遗传学特征进行分析,并应用外显子组测序技术对其致聋基因进行初步筛查及分析,报告如下。
1 资料与方法
1.1 家系调查方法 本研究家系的调查分析已获得解放军总医院伦理委员会的伦理认证及认可。该家系(编号:HB-Z177)位于河北省,先证者于2010年6月就诊于解放军总医院耳鼻咽喉头颈外科门诊,分别进行了纯音测听、声导抗、听性脑干反应、DPOAE及颞骨CT扫描检查,结果提示为双侧对称的中度至重度感音神经性聋,呈进行性加重,听力曲线呈下降型,该家系成员无明确的耳毒性药物应用史及噪声接触史,不伴其他器官系统异常,诊断为遗传性感音神经性聋。2011年4月对该家系成员进行了问卷调查并签署知情同意书,对所有受检者进行了详细的病史询问、严格的专科检查及听力学检测,均采集5~10ml外周静脉血,用于后续的基因组DNA提取和遗传学特征研究。
1.2 耳聋表型判断标准 根据2003年9月Van Camp等[17]提议的《关于非综合征型遗传性聋家系遗传学及听力学描述术语建议案》,依据是否伴其他器官或系统异常,分为非综合征型和综合征型聋;按照耳聋出现的时间分为语前聋、语后聋;依据听力较好耳0.5~4kHz的平均听阈将听力损失程度分为四级,轻度听力损失为20~40dB HL,中度听力损失为41~70dB HL,重度听力损失为71~95dB HL,极重度听力损失为>95dB HL。
1.3 全基因外显子组测序 运用ILLUMINA二代测序平台进行家系全基因外显子组测序,选取先证者V2基因组DNA 3~5μg完成建库后上机测序,并对测序结果进行基本的数据分析,同时从获得的测序数据中选择高频单核苷酸变异(SNV)对结果进行验证,以确认二代测序数据的准确性。
1.4 数据分析 统计外显子组样品测序数据生成量以及测序深度和覆盖度等数据。初步查阅拟筛查的变异:①杂合子;②排除千人基因组计划和寡核苷酸多态性数据库(database of single nucleotide polymorphism,dbSNP数据库)中已报道的出现频率≥0.001的SNP及短串联重复序列(short tandem repeats,STR)变异;③与已有的耳聋基因数据库进行比较,优先筛查已知突变位点。排除已知变异后,参考以往文献对候选基因的阐述,筛查未知变异,针对测序发现的SNV及碱基插入或缺失进行质控分析,结合家系常染色体显性遗传特征,去除纯合状态的变异及冗余数据,优先考虑有重要功能意义的变异;参考用于预测氨基酸改变是否影响其蛋白功能的点突变预测(sorting intolerant from tolerant,SIFT)分值(http://sift.jcvi.org/www/SIFT_chr_coords_submit.html)和用于预测非同义突变造成的氨基酸改变是否影响蛋白功能的多态性表型(polymorphism phenotyping,POLYPHEN)分 值(http://genetics.bwh.harvard.edu/pph/pph_help_text.html);选取在脑或周围神经系统有表达或对应的分子功能有重要意义的基因变异优先进行筛查。
1.5 Sanger测序验证 针对突变位点设计引物,对该家系患者基因组DNA进行PCR扩增和Sanger测序,必要时对家系正常成员进行对照验证。
2 结果
2.1 HB-Z177家系系普图和听力学特征 该家系系谱图见图1(正常成员的后代未画出,也未纳入统计),HB-Z177家系共五代合计27人(男13人,女14人),现存19人,主要为第三代至第五代成员,年龄介于15~60岁之间,共10人患双耳感音神经性聋,其中表型不确定者1人,现存确诊患者7人。因时间久远,已无法确定第一代患者性别;第二代共4名男性,患病2人,患病成员与正常成员比例为1:1;第三代1男3女共4人,有1男2女共3人患病,患病成员与正常成员比例为3:1,此比例偏离1:1,与该代成员数偏少有关;第四代4男5女共9人,有1男3女共4人患病,1人(IV11)表型不明确,患病成员与正常成员比例接近1:1;第五代共8人,1人患病,部分成员可能未到发病年龄。先证者(V2)26岁,于12岁左右开始出现听力下降伴耳鸣,纯音听阈测试显示双耳对称性中度至重度感音神经性聋,听阈曲线呈下降型(图2f)。其余患者发病年龄不等,10~30岁出现听力下降,且年龄均已超过45岁,均表现为双耳对称性进行性加重的重度感音神经性聋,听阈曲线双耳对称均为平坦型,第III7、IV1、IV3、IV5、IV7患者的纯音听阈图见图2a~e。由此可见,HB-Z177家系符合常染色体显性遗传性耳聋特征。
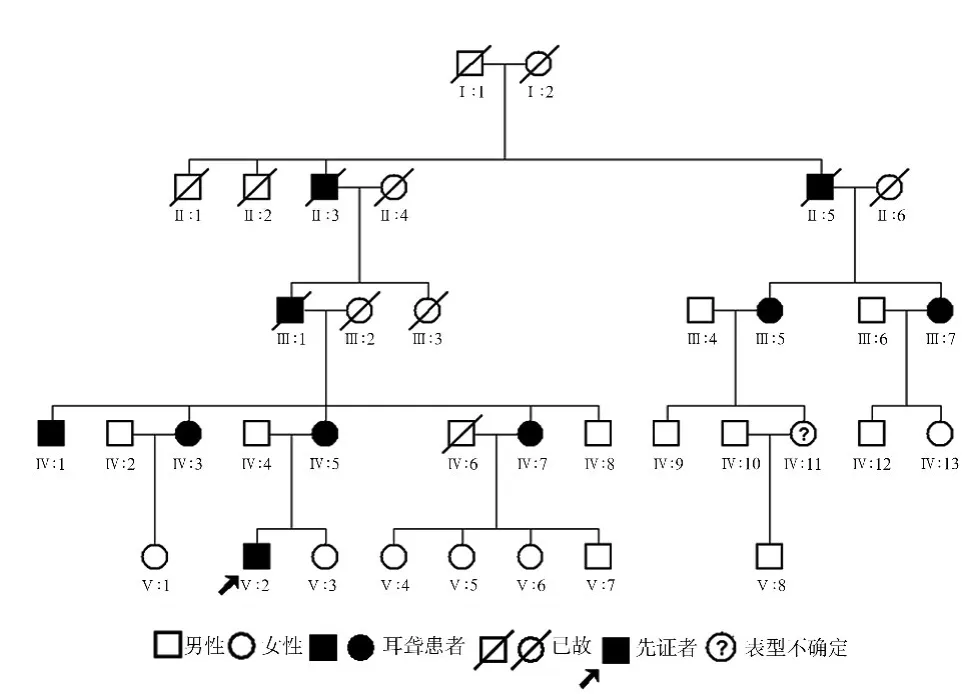
图1 HB-Z177家系系谱图
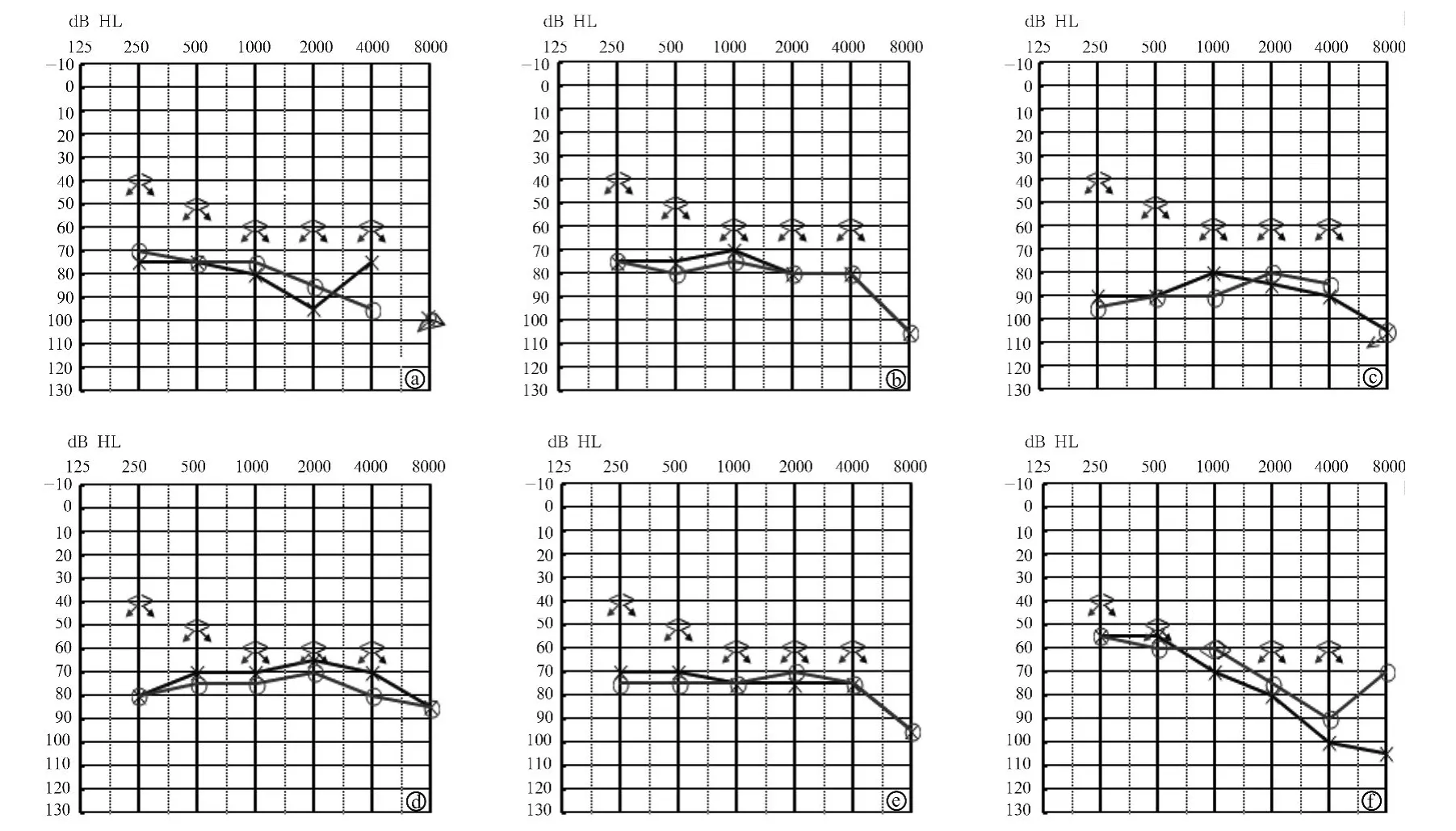
图2 HB-Z177家系患者听力图
2.2 全外显子组测序分析结果 总览全外显子组测序数据库,平均测序深度及覆盖度基本符合测序要求。单杂合的变异总数为936个,筛查到的已知耳聋基因的突变位点共6个(表1),根据上述筛选标准,因USH2A(rs3745079)在千人基因组中出现的频率为0.037(≥0.001),TMIE(rs10578999)及DIAPH1(rs35249023)为STR,故均予以直接剔除;待筛查的已知致聋基因变异位点共3个,分别为EYA4(NM_004100:c.1486A>T)、MYO7A(NM_000260:c.3602G>C)和 ESPN(NM_031475:c.2225C>T)。剩余的可纳入优先考虑的候选SNV共234个,根据上述排除已知变异后的筛选标准,仅选取其中4个未知基因变异位点(表1)DNMT1(NM _001130823:c.4381C>T)、BMP2(NM _001200:c.393A>T)、TP63(NM_001114979:c.854G>A)及 RAI1(NM_030665:c.4162G>A)进行Sanger测序验证。
2.3 Sanger测序结果 选取家系其余5名患病成员(III7,IV1,IV3,IV5,IV 7)及1名正常成员(IV8)应用Sanger测序验证,针对WES的初步筛选结果,依次完成了已知基因EYA4(NM_004100:c.1486A>T)、MYO7A(NM_000260:c.3602G>C)和 ESPN(NM_031475:c.2225C>T)的Sanger测序验证,没有发现这些碱基改变与临床表型有共分离现象,予以排除。此筛查结果提示HB-Z177家系的遗传性聋致病基因可能并非已知耳聋基因,而是由未知基因变异引起。后续对已知耳聋基因以外的DNMT1(NM_001130823:c.4381C>T)、BMP2(NM_001200:c.393A>T)、TP63(NM_001114979:c.854G>A)及 RAI1(NM_030665:c.4162G>A)进行了筛查,这些变异位点亦未与临床表型共分离,亦予以排除。
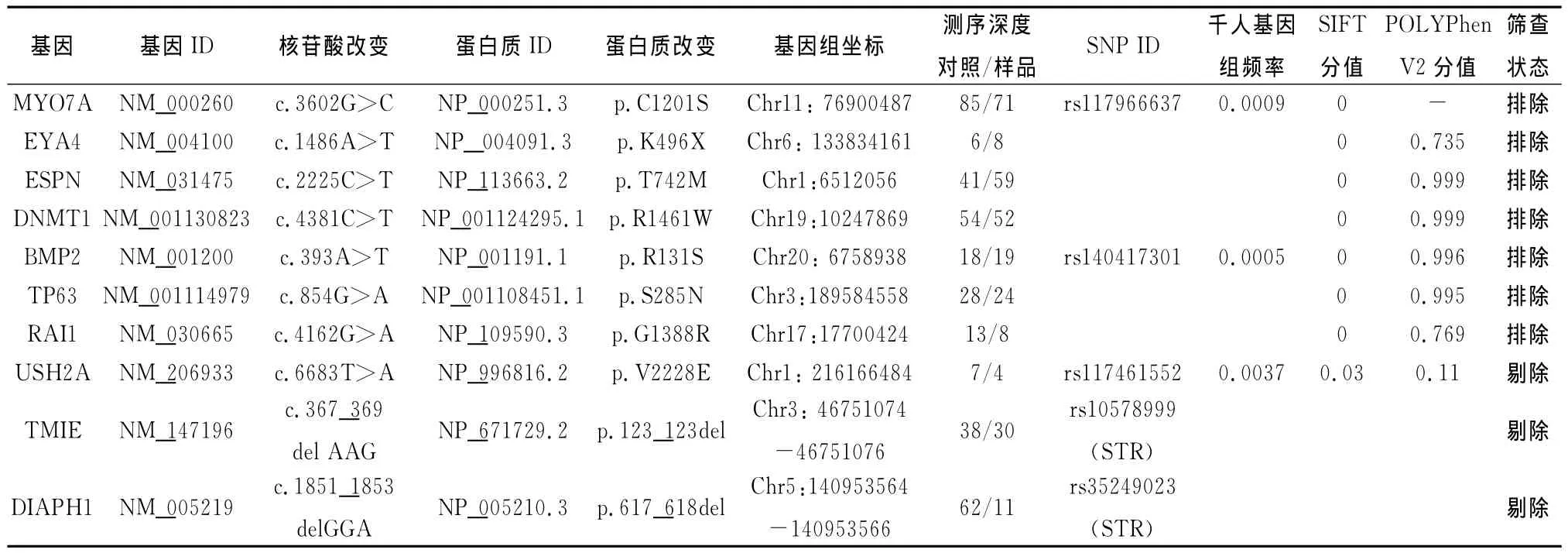
表1 全外显子组测序的6个已知耳聋基因突变位点及4个未知基因可疑变异的数据信息及筛查结果
3 讨论
从文中结果看,HB-Z177家系第五代先证者V2与其他患病成员(III7、IV1、IV3、IV5、IV7)年龄差较大(V2与第三、四代患病成员年龄最小者IV7亦相差19岁),而V2现26岁,于12岁发病,表现为双耳中重度感音神经性聋,听阈曲线呈下降型,其他患病成员均于10~30岁发病,现均已超过45岁,均表现为双耳重度感音神经性聋,听阈曲线为平坦型,故仅能推测该家系耳聋发病初期可能以高频听力下降为主,是否随年龄增长而进行性加重且逐渐累积全频尚不可知;综合其他患病成员的信息可明确,年龄超过45岁后,患者的听阈曲线以平坦型为主,均表现为重度感音神经性聋。该家系患病成员的发病年龄、听力进行性加重的特点和均一对称的听阈曲线均提示其为遗传性聋。从HB-Z177家系图可知,该家系连续三代以上有成员发病,患病成员中5男5女,男女发病比例相等;第二代至第四代成员均达到发病年龄,除成员IV11外,表型均已明确,患者与正常人比例为9:7,患病成员与正常人比例接近,因第五代成员年龄介于15到26岁之间,可能有部分成员未到发病年龄而未出现表型,故此代患病成员与正常人比例为1:7,偏离1:1;正常成员后代均正常,患病成员父母均有1人患病。结合以上遗传学分析可知,HB-Z177家系符合常染色体显性遗传非综合征型感音神经性聋特点。
千人基因组计划为人们提供了丰富的人类遗传变异数据[18],为新一代测序技术的数据分析工作奠定了基础。外显子组测序高效、快捷,费用正逐步降低,且可实现目标区域的高覆盖度,鉴于大多数的致病性变异主要位于编码区或内含子与外显子相邻区域,外显子组测序已被广泛地应用到孟德尔遗传性疾病研究中,大大地提升了变异的检测效率。应用WES研究家族遗传性疾病,更有助于致病基因的快速筛选。HB-Z177家系仅选取1个样本进行外显子组测序,得到的数据量庞大,后期严谨的数据分析工作对于致病基因的鉴定尤为重要。本家系为常染色体显性遗传,致病基因应为杂合状态,故剔除数据库中的纯合性变异;在所有的杂合性突变中,将已知的或已定位的致聋基因作为重点筛查对象,但剔除千人基因组计划数据库、dbSNP数据库已报道的出现频率≥0.001的SNP及STR区域的变异,USH2A为Usher综合征的已知致病基因,c.6683T>A(NM_206933)为 已 知 SNP(rs117461552),且在千人基因组中的频率为0.0037(≥0.001),POLYPhen V2 分值较低,提示该变异对蛋白功能影响不大,暂不予以考虑;位于STR区域的TMIE(rs10578999)在千人基因组数据库中,已明确其为非致病性变异;DIAPH1(rs35249023)亦位于STR,因STR区域较少出现致病性突变,故暂不考虑其致病的可能性,均予以剔除。对筛选出的3个已知致聋基因变异进行Sanger测序验证,因没有发现突变位点与临床表型有共分离现象而予以全部排除。除以上所检测的已知耳聋基因外,数据库中未见其他杂合状态的已知耳聋基因变异,此筛查结果提示该家系的致聋基因可能为未知基因。继续按照以上的筛查策略,选择在脑或周围神经系统等部位有表达或有重要功能的基因DNMT1、BMP2和TP63的对应突变位点继续筛查,以期找到家系的新致聋基因,但因碱基变异与耳聋表型未出现共分离现象予以排除。
本研究结果显示,HB-Z177家系符合常染色体显性遗传规律,听力学表现为双侧对称的中度至重度感音神经性聋。对外显子组测序发现的3个已知耳聋基因突变位点及4个可疑的变异位点在家系其他成员进行Sanger测序验证后,未发现这些突变碱基位点与耳聋表型共分离,提示其致聋基因可能为未知的新的致聋基因。本研究中,因单个样品测序产生的基因数据信息量庞大,基因筛选工作的力度和难度较大,不利于致病基因的快速筛选,故拟进一步增加1~2个样品行全外显子组测序,以便快速鉴定出该家系的致聋基因。
1 Bitner-Glindziez M.Hereditary deafness and phenotyping in humans[J].Br Med Bull,2002,63:73.
2 Cryns K,Van Camp G.Deafness genes and their diagnostic applications[J].Audiol Neurootol,2004,9:2.
3 Zhao Y,Zhao F,Zong L,et al.Exome sequencing and linkage analysis identified tenascin-C(TNC)as a novel causative gene in nonsyndromic hearing loss[J].PLoS One,2013,8:e69 549.
4 Girotto G,Abdulhadi K,Buniello A,et al.Linkage study and exome sequencing identify a BDP1mutation associated with hereditary hearing loss[J].PLoS One,2013,8:e80 323.
5 Jaworek TJ,Richard EM,Ivanova AA,et al.An alteration in ELMOD3,an Arl2GTPase-activating protein,is associated with hearing impairment in humans[J].PLoS Genet,2013,9:e1003 774.
6 Yariz KO,Duman D,Seco CZ,et al.Mutations in OTOGL,encoding the inner ear protein otogelin-like,cause moderate sensorineural hearing loss[J].Am J Hum Genet,2012,91:872.
7 Delmaghani S,Aghaie A,Michalski N,et al.Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98profound deafness[J].Hum Mol Genet,2012 ,21:3 835.
8 Santos-Cortez RL1Lee KAzeem Zet al.Mutations in KARS,encoding lysyl-tRNA synthetase,cause autosomalrecessive nonsyndromic hearing impairment DFNB89[J].Am J Hum Genet,2013,93:132.
9 Rehman AU,Santos-Cortez RL,Morell RJ,et al.Mutations in TBC1D24,agene associated with epilepsy,also cause nonsyndromic deafness DFNB86[J].Am J Hum Genet,2014,94:144.
10 Walsh T,Shahin H,Elkan-Miller T,et al.Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2as the cause of nonsyndromic hearing loss DFNB82[J].Am J Hum Genet,2010,87:90.
11 Rehman AU,Morell RJ,Belyantseva IA,et al.Targeted capture and next-generation sequencing identifies C9orf75,encoding taperin,as the mutated gene in nonsyndromic deafness DFNB79[J].Am J Hum Genet,2010,86:378.
12 Schraders M,Haas SA,Weegerink NJ,et al.Next-generation sequencing identifies mutations of SMPX,which encodes the small muscle protein,X-linked,as a cause of progressive hearing impairment[J].Am J Hum Genet,2011,88:628.
13 Rost S,Bach E,Neuner C,et al.Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6[J].Eur J Hum Genet,2014,22:208.
14 Pierce SB,Walsh T,Chisholm KM,et al.Mutations in the DBP-deficiency protein HSD17B4cause ovarian dysgenesis,hearing loss,and ataxia of Perrault syndrome[J].Am J Hum Genet,2010 ,87:282.
15 Pierce SB,Gersak K,Michaelson-Cohen R,et al.Mutations in LARS2,encoding mitochondrial leucyl-tRNA synthetase,lead to premature ovarian failure and hearing loss in Perrault syndrome[J].Am J Hum Genet,2013,92:614.
16 Jenkinson EM,Rehman AU,Walsh T,et al.Perrault syndrome is caused by recessive mutations in CLPP,encoding a mitochondrial ATP-dependent chambered protease[J].Am J Hum Genet,2013,92:605.
17 王秋菊,译.关于非综合征型遗传性听损伤家系遗传学及听力学描述术语建议案[J].中华耳科学杂志,2003,1:46.
18 Abecasis GR,Altshuler D,Auton A,et al.A map of human genome variation from population scale sequencing[J].Nature,2010,467:1 061.
猜你喜欢
中国现代医生(2022年19期)2022-11-04
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
郑州大学学报(医学版)(2015年2期)2015-02-27
湖北农业科学(2014年11期)2014-09-10
