
人感染H6N1禽流感病毒HA基因的分子特征及进化分析
2014-11-20 11:41杨建课高继光林爱琴徐思斌
中国人兽共患病学报 2014年8期
杨建课,宫 磊,高继光,林爱琴,徐思斌
2.皖南医学院医学遗传学研究室;
Email:ajiankebc@126.com
A型禽流感病毒属于正粘病毒科流感病毒属,在全世界范围内的家禽、野鸟、哺乳动物及人类中广泛传播[1]。该病毒基因组拥有8条负RNA链,至少编码11种不同功能的蛋白质。依据其表面血凝素(hemagglutinin,HA)和神经氨酸酶(neuraminidase,NA)的抗原性,将其分成16个HA亚型和9个NA亚型,最近又在一些蝙蝠体内分离并鉴定出了新的H17N10亚型[2]。这些病毒对人类的生活和健康都构成了极大的威胁。
虽然人们早已分离出低致病性禽流感病毒(LPAI)H6N1,且研究表明其在一定条件下具有在人和哺乳动物间交叉感染的能力[3-4],但很少见到人感染的病例。2013年6月,台湾发现了首例人感染H6N1禽流感病例[5-7],引人关注。特拟对人感染H6N1禽流感病毒的HA基因及其近缘序列的分子特征及进化分析,以探求其变异规律、致病机制、来源及进化情况,为禽流感病毒的防治提供理论依据。
1 材料与方法
1.1 数据来源 通过Blast方法从美国国立生物技术信息中心(NCBI)流感病毒资源库(Flu)和全球禽流感基因共享数据库(GISAID)中获得人感染H6N1禽流感病毒HA基因及近缘序列共47条,其中42条来自台湾(如表1)。经 MAFFT version 7.110比对和BioEdit version 7.2.1人工校正后作为分析数据集。
1.2 系统发育树重建 分别采用最大似然法(ML)、最大简约法(MP)、贝叶斯推理法(BI),以A/CK/TW/G23/87(DQ376692)为外类群,jModelTest version 2.1.4的AIC标准计算的GTR+G为最适模型,重建其系统发育树。其中ML、MP树分别在软件 PhyML3.0、PAUP version 4.0b10中运算执行,自举检验值(Bootstrap)1 000次重复检验;BI树由MrBayes 3.2运算获得,4个独立的马可夫链-蒙特卡罗(MCMC)数据模拟技术运算1百万代,每100代取样一次,舍弃前2 500个老化样本(burnin)后,其余样本用来推测后验概率。
1.3 分子特征分析 分别通过NetNGlyc 1.0、NetOglyc 4.0预测氨基酸序列的N-糖基化位点和O-糖基化位点。以人感染H6N1禽流感病毒的HA蛋白序列为参考,分析其变异位点和裂解位点的特征,并通过 ProtParam、DiANNA 1.1、SignalP 4.1分别预测其理化性质、二硫键和信号肽的位置。
1.4 进化速率和分歧时间推断 采用GTR+G模型、贝叶斯轮廓式(Bayesian Skyline)种群增长模式和对数正态分布松弛分子钟模型(CoV>0),通过BEAST v1.7.5推测42条台湾数据的进化速率(位点/年)和最近共同祖先(TMRCA)时间。2个独立的MCMC数据模拟技术运算5千万代。FigTree v1.4.0显示树的拓扑结构和相关信息。
2 结 果
2.1 系统发育分析 为更准确分析人感染H6N1禽流感病毒HA基因的来源和进化历程,我们分别构建了ML、MP和BI树,三种树的拓扑结构基本一致,仅支持率略有不同(如图1)。根据树的拓扑结构、支持率、病毒出现的时间、地点和序列特征等分析,由A/CK/TW/G23/87进化而来的46株病毒聚为两支。支1所含毒株均来自台湾,支持率 ML/MP/BI分别为99/99/1,其内部进一步聚成三个亚支,其中支1.3支持率为99/99/1,包括 A/CK/TW/PF3/02及1999至2004年间分离的11株病毒;支1.2支持率为100/100/1,共4株病毒,其中人感染 H6N1禽流感病毒(A/TW/2/13)与A/CK/TW/A2837/13亲缘关系最近,互为姐妹支,再与2004和2005年的两株禽感染病毒聚在一起;支1.1的支持率为85/86/0.96,含有2001至2010年间分离的20株病毒,毒株数最多。支1.1和1.2互为姐妹 支,再 与 A/CK/TW/1205/01 和 A/CK/TW/0329/01聚在一起,支持率分别为88/86/0.88和100/100/1。支2支持率为81/76/1,含5株来自广西和荷兰的禽流感病毒,寄主全为水禽,距根部最近。
2.2 HA蛋白的分子特征 人感染H6N1病毒的HA蛋白的分子量约为63.6KDa,等电点为6.07,由567个氨基酸残基组成,其中亮氨酸(L)含量最高(8.3%),组氨酸(H)最少(1.6%)。1至16位为信号肽序列,引导新合成的HA蛋白进入内质网。序列共有15个半胱氨酸(C),可形成7个二硫键(6与92、44与479、57与137、279与552、283与468、307与475、542与549);含有5个 N-糖基化位点(第13、25、169、293、544位)和2个 O-糖基化位点(第130、134位)(如表1),其中N-糖基化位点在不同支系间较为保守,O-糖基化位点变异较大(支2多为一个、支1.3多为三个,支1.1和1.2多为两个)。HA1和HA2的裂解位点位于第331和332位间,其上游序列PQIATR,较为保守,仅含有一个碱性氨基酸,其中裂解位点上游第三位氨基酸(即329位)变异较大,较早分离的几株病毒为赖氨酸(K),而支1.2的4株病毒为特有的丙氨酸(A),其余毒株为谷氨酸(E)。虽然人感染H6N1禽流感病毒的HA蛋白序列第193N、226Q、228S与其它禽感染病毒一致,但P186L、A287T为其所特有。此外,55K、100L、157D、160T、200K、238R、480M、495K为毒株 A/TW/2/13和 A/CK/TW/A2837/13特有,86N、273D、329A为支1.2所特有。
2.3 进化速率和TMRCA时间推测对来自台湾的42条HA基因进行了进化速率(位点/年)和TMRCA推测,结果如表2。节点C进化速率最大,为5.99×10-3位点/年,说明支1.2的进化速速最快;节点F次之,为5.90×10-3位点/年;而节点B进化速率最小,为4.40×10-3位点/年,说明支1.3进化的最慢。节点D所代表的最近共同祖先时间为2012年,故推测人感染H6N1禽流感病毒的HA基因于2012年早期由家禽的序列进化而来。此外,C、D节点间枝长较长,TMRCA相差10年之久。

表1 人感染H6N1及近缘禽流感病毒HA基因的登录号及糖基化位点(参考H3序列位置)Tab.1 Accession numbers and glycosylation sites of the HA gene from human-infecting H6N1and allied avian influenza virus(H3 numbering)
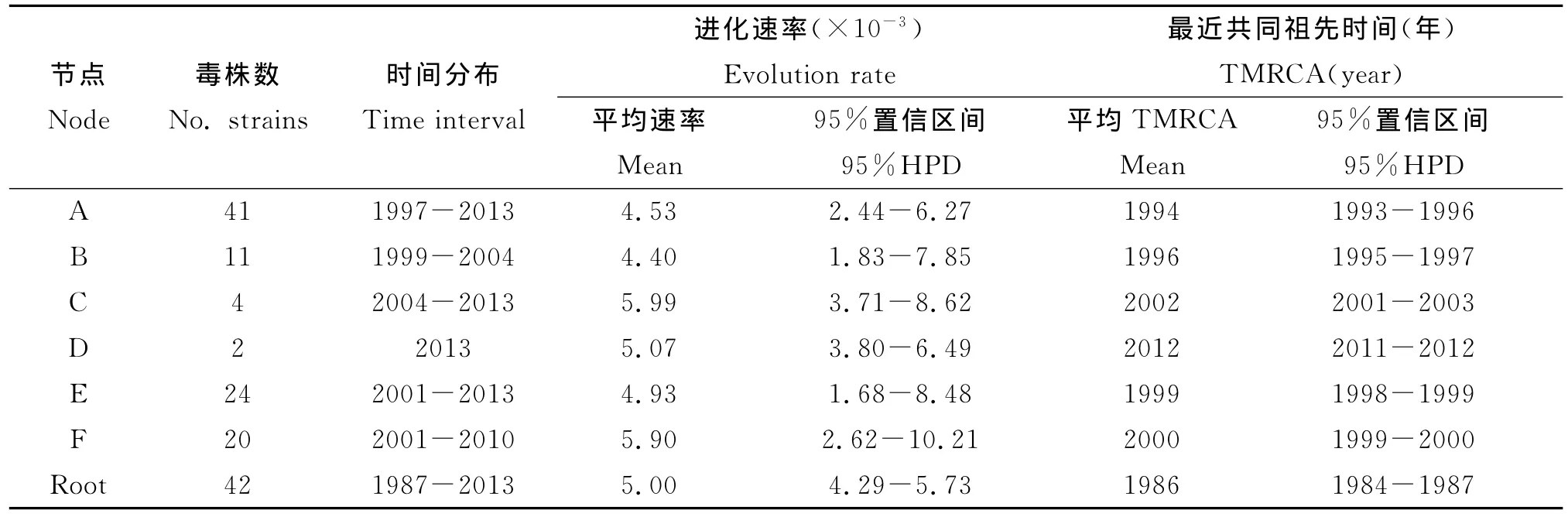
表2 不同节点的进化速率(位点/年)和最近共同祖先(TMRCA)时间Tab.2 Substitution rates(sites/year)and the most recent common ancestor(TMRCA)of some significant nodes
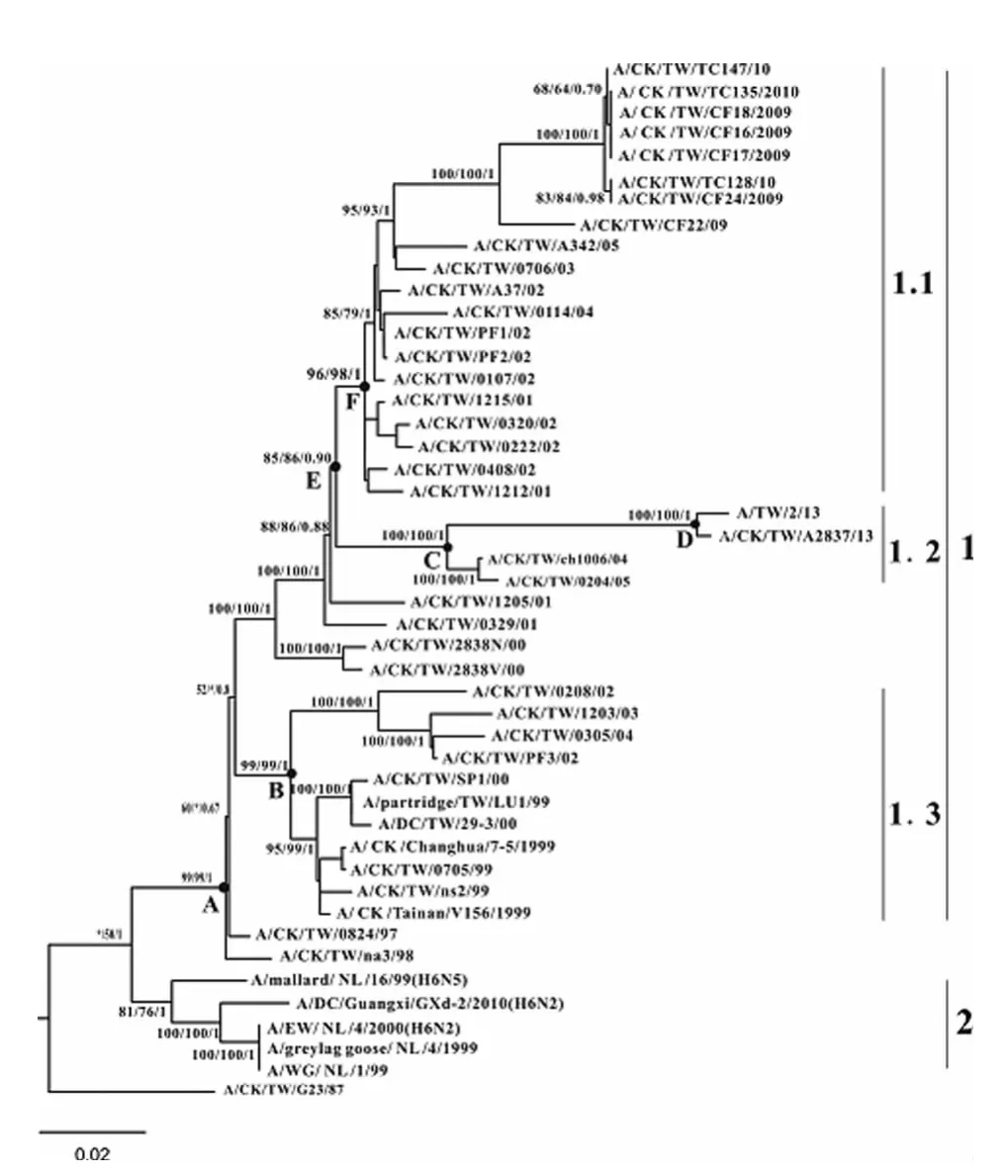
图1 人感染H6N1及近缘禽流感病毒HA基因的系统发育树图。支上数字分别表示 ML、MP、BI树的支持率,大写的英文字母代表一些重要的节点,加粗数字表示不同的支。Fig.1 Phylogenetic trees of the HA genes from human-infecting H6N1and allied avian influenza virus by maximum likelihood(ML),maximum parsimony(MP)and Bayesian inference(BI)
3 讨 论
虽然相对于高致病性的H7N9[8],低致病性的H6N1对人的危害性较小,但其对家禽的孵化率、死亡率及产蛋量等影响很大[4],且可能与其它禽流感病毒重配产生新型高致病性流感病毒,如A/Hong Kong/156/97(H5N1)的产生[9],应高度关注。截止目前,由 A/CK/TW/G23/87进化而来的 H6亚型至少有2种类型——支1和2,支2病毒主要出现在欧洲和中国的南部,支1病毒全分布在台湾。其中亚支1.3所含病毒出现时间相对较早,进化速率小,可能已不是优势种群,甚至已完全消失。而亚支1.1、1.2病毒可能为当地优势种群,正在流行且进化速度快,说明其还在不断的进化和适应环境。人感染H6N1禽流感病毒HA基因属于亚支1.2,高的支持率和一些特有的氨基酸位点,都表明这是早在2002年就进化出的新类型,并已适应外部环境。然而目前分离的毒株并不多,可能因为其低致病性,很多毒株未被发现,节点C、D间的长枝也说明如此[7]。Wei等[6]研究认为,人感染H6N1禽流感病毒HA基因可能由A/CK/TW/PF3/02,经A/CK/TW/0204/05进化而来,而我们的结果不完全支持此观点。支1.1和1.2先与A/CK/TW/1205/01、A/CK/TW/0329/01聚在一起后再与A/CK/TW/PF3/02所在的支1.3互为姐妹支,结合有关数据[5,7,10]可见:人感染H6N1禽流感病毒HA基因最早由A/CK/TW/G23/87,经A/CK/TW/0824/97、A/CK/TW/1205/01、A/CK/TW/0204/05,直到2012年由家禽的序列进化而产生,与A/CK/TW/PF3/02(H6N1)病毒为共进化关系。
HA蛋白以同源三聚体的形式镶嵌在病毒表面,作为主要的表面抗原,与病毒复制、毒力及宿主特异性等密切相关。研究表明,裂解位点上游碱性氨基酸的数目决定着HA蛋白能否被裂解成HA1和HA2,进而影响病毒的复制和毒力[11-12]。人感染H6N1禽流感病毒HA蛋白的裂解位点上游6个氨基酸里仅有1个碱性氨基酸,表明其仍是低致病性的[6-7],但位点328、329(裂解位点上游第4、第3位)具有很大的变异性,可见其受到很大的选择压力,将来有可能进化产生高致病性的人感染H6亚型流感病毒。前人研究还表明HA蛋白的第226位(参照H3序列)如为Q,则特异性地与禽类含唾液酸α-2,3半乳糖的糖蛋白受体结合;如为L,则特异性地与人类含唾液酸α-2,6半乳糖的糖蛋白受体结合;此外,193R、228G、糖基化位点等也与宿主的特异性有关[11,13-14]。人感染H6N1禽流感病毒H蛋白193N、226Q、228S未表现出人感染型的特性,可能尚未来得及进化,但特有的P186L也许是其感染人的原因[7,13]。同时,特有的A287T在病毒宿主的选择上是否具有一定作用,目前还不清楚。该蛋白拥有5个N-糖基化位点,其中4个位于HA1区,1个位于HA2区,5个位点都比较保守,表明其具有重要的生物学功能[15]。同时拥有2个O-糖基化位点,具体功能未知,但在不同类群间有所变异,这可能是适应性进化的结果。
综上,人感染H6N1禽流感病毒的HA基因由家禽的进化而来,并具备了感染人的能力,由于其高的进化速率和病毒重配的可能性,加以时日有可能进化出新型高致病性的H6亚型,人们应高度重视,并能及时的给与监测和防治。
[1]Zhou H,Jin M,Chen H,et al.Genome-sequenee analysis of the pathogenic H5N1avian influenza A virus isolated in china in 2004[J].Virus Genes,2006,32(1):85-95.DOI:10.1007/s11262-005-5849-9
[2]Tong S,Li Y,Rivailler P,et al.A distinct lineage of influenza A virus from bats[J].Proc Natl Acad Sci U S A,2012,109(11):4269-4274.DOI:10.1073/pnas.1116200109
[3]Beare A,Webster R.Replication of avian influenza viruses in humans[J].Arch Virol,1991,119(1-2):37-42.DOI:10.1007/BF01314321
[4]Lee MS,Chang PC,Shien JH,et al.Genetic and pathogenic characterization of H6N1avian influenza viruses isolated in Taiwan between 1972and 2005[J].Avian Dis,2006,50(4):561-571.DOI:10.1637/7640-050106R.1
[5]Yuan J,Zhang L,Kan X,et al.Origin and molecular characteristics of a novel 2013avian influenza A(H6N1)virus causing human infection in Taiwan[J].Clin Infect Dis,2013,57(9):1367-1368.DOI:10.1093/cid/cit479
[6]Wei SH,Yang JR,Wu HS,et al.Human infection with avian influenza A H6N1virus:an epidemiological analysis[J].Lancet Respir Med,2013,1(10):771-778.DOI:10.1016/S2213-2600(13)70221-2
[7]Shi W,Shi Y,Wu Y,et al.Origin and molecular characterization of the human-infecting H6N1influenza virus in Taiwan[J].Protein Cell,2013,4(11):846-853.DOI:10.1007/s13238-013-3083-0
[8]Liu D,Shi W,Shi Y,et al.Origin and diversity of novel avian influenza A H7N9viruses causing human infection:phylogenetic,structural,and coalescent analyses[J].Lancet,2013,381(9881):1926-1932.DOI:10.1016/S0140-6736(13)60938-1
[9]Chin P,Hoffmann E,Webby R,et al.Molecular evolution of H6influenza viruses from poultry in Southeastern China:prevalence of H6N1influenza viruses possessing seven A/Hong Kong/156/97(H5N1)-like genes in poultry[J].J Virol,2002,76(2):507-516.DOI:10.1128/JVI.76.2.507-516.2002
[10]Zhang L,Zhang Z.The origin of human-infecting avian influenza A H6N1virus[J].bioRxiv,2013,in press.DOI:10.1101/000398
[11]Schrauwen EJA,Graaf M,Herfst S,et al.Determinants of virulence of influenza A virus[J].Eur J Clin Microbiol Infect Dis,2014,33(4):479-490.DOI:10.1007/s10096-013-1984-8
[12]Subbarao K,Chen H,Swayne D,et al.Evaluation of a genetically modified reassortant H5N1influenza A virus vaccine candidate generated by plasmid-based reverse genetics[J].Virology,2003,305(1):192-200.DOI:10.1006/viro.2002.1742
[13]Xiong X,Martin SR,Haire LF,et al.Receptor binding by an H7N9influenza virus from humans[J].Nature,2013,499(7459):496-499.DOI:10.1038/nature12372
[14]Hatta M,Gao P,Halfmann P,et al.Molecular basis for high virulence of Hong Kong H5N1influenza A viruses[J].Science,2001,293(5536):1840-1842.DOI:10.1126/science.1062882
[15]Chen W,Zhong Y,Qin Y,et al.The evolutionary pattern of glycosylation dites in influenza virus(H5N1)hemagglutinin and neuraminidase[J].PLoS One,2012,7(11):e49224.DOI:10.1371/journal.pone.0049224
猜你喜欢
环球时报(2022-10-10)2022-10-10
复旦学报(医学版)(2021年5期)2021-10-13
癌症进展(2018年11期)2018-12-30
中成药(2017年12期)2018-01-19
小康(2017年25期)2017-09-07
中国畜牧兽医文摘(2015年9期)2015-12-29
中国畜牧兽医文摘(2015年9期)2015-12-29
医学研究杂志(2015年7期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
