
一个非综合征性耳聋家系的GJB2基因突变分析及产前诊断
2014-11-05 01:19陈晓华余小艳周洁姜威胡俊钟山杨国华
解放军医学杂志 2014年7期
陈晓华,余小艳,周洁,姜威,胡俊,钟山 ,杨国华
耳聋出生缺陷是影响我国出生人口素质的重要问题之一[1],2006年12月第二次全国残疾人抽样调查结果显示,耳聋位居我国各类残疾之首,听力残疾者高达2780万,且每年以3万聋儿的新发速度在增长[2],约60%的先天性耳聋由遗传性因素导致[3]。非综合征性耳聋(non-syndromic hearing impairment,NSHI)占遗传性耳聋的70%,约77%的NSHI为常染色体隐性遗传[4-5]。缝隙连接蛋白2(gap junction protein beta-2,GJB2)基因突变是常染色体隐性遗传听力损失的主要原因之一[6],约有50%的常染色体隐性遗传NSHI患者存在该基因突变[7]。目前已知的与耳聋有关的GJB2基因突变位点有100多个,其中235delC突变在东亚人群发生频率较高[8]。在耳聋基因诊断的临床工作中,我们发现一个同时携带GJB2 235delC和176-191del16bp的家系,对此家系相关成员的基因型进行了分析,并对孕妇进行产前诊断。
1 资料与方法
1.1 研究对象 样本取自湖北孝感市中心医院妇产科。孕妇(Ι-2)33岁,身体健康,受检时妊娠20周,孕期常规体检未见异常,其第一胎为先证者(Ⅱ-1),男,7岁,汉族,智力正常,出生后即无听力、无语言能力,心、脑、肾等器官无异常。孕妇丈夫(Ι-1)32岁,身体健康。夫妇二人已育有一子,即为耳聋先证者(Ⅱ-1),第二胎(Ⅱ-2)胎龄20周,家系谱见图1。
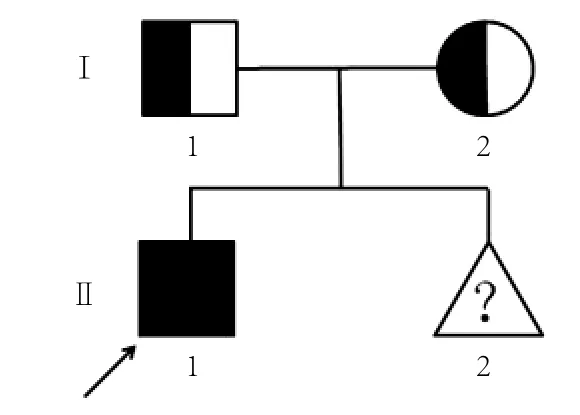
图1 患者家系图Fig. 1 Pedigree of the non-syndromic deafness family
1.2 研究方法
1.2.1 临床听力学检测 对先证者进行电耳镜检查、纯音测听、声导抗检测及颞骨CT扫描检查。以2003年《关于非综合征型遗传性听损伤家系遗传学及听力学描述术语建议案》为耳聋诊断标准,耳聋程度按照两耳中听力较好耳的平均听阈(0.5~4.0kHz听阈的平均值)来评估: ≤20dB HL为正常;20~40dB HL为轻度听力损失;41~70dB HL为中度听力损失;71~95dB HL为重度听力损失;>95dB HL为极重度听力损失。
1.2.2 家系所有成员GJB2基因型测序及比对
1.2.2.1 基因组DNA抽提 取得该家系成员书面同意后,抽取该家系成员Ι-1、Ι-2、Ⅱ-1外周抗凝血各2ml,应用蛋白酶K法提取白细胞基因组DNA;超声引导下行羊膜腔穿刺术抽取孕妇羊水20ml,无菌培养羊水细胞1周后,应用蛋白酶K法提取羊水细胞基因组DNA。紫外分光光度计进行DNA定量与纯度分析,4℃保存备用。
1.2.2.2 PCR扩增GJB2基因 通过NCBI GenBank查询到GJB2编码区序列,利用Primer 5软件设计引物。上游引物:5'-TTGGTGTTTGCTCAGGAAGA-3',下游引物:5'-GGCCTACAGGGGTTTCAAAT-3',产物大小为960bp。应用ABI 9700热循环仪进行DNA扩增。反应体系为:10×Buffer 2.5 μl,dNTP 2.5μl,Taq酶2μl,10μmol/L上下游引物各1 μl,100ng DNA模版1 μl,ddH2O 15μl。反应条件:94℃预变性5min;94℃变性30s、55℃退火30s、72℃延伸1min,35个循环;72℃延长10min。对扩增后产物行1.5%琼脂糖凝胶电泳粗测,结果显示家系中所有成员均含有GJB2基因(图2)。
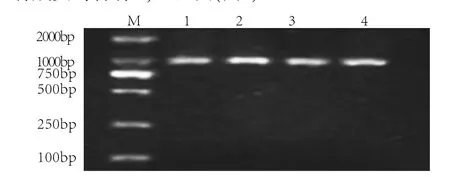
图2 家系各成员GJB2基因PCR电泳图Fig.2 GJB2 electrophoresis analysis of the family members
1.2.2.3 PCR产物测序及TA克隆后测序 将PCR扩增产物用快速DNA产物纯化试剂盒(北京康为世纪生物科技有限公司)进行纯化,送上海生工(Sangon)生物工程股份有限公司测序。采用上、下游引物双向测序证实的序列变异认定为突变。针对测序图出现的难辨杂峰,对扩增产物进行TA克隆后再挑取单克隆进行测序:将PCR纯化后的扩增产物连接到pMDI8-T载体,连接产物转化大肠埃希菌DH5α,每一产物挑取多个克隆菌落进行培养,收集菌液,抽提质粒进行测序。在多个克隆中同时存在的序列变异认定为突变。
1.3 序列比对 应用Bioedit软件将测序结果与NCBI网站提供的人类GJB2基因标准序列(GenBank:AY280971.1;GI:33391196)进行比对。利用Omiga 2.0软件对核酸序列进行分析。
2 结 果
2.1 听力学检测结果 纯音电测听检查结果显示,先证者左右耳听力均大于71分贝,属于重度耳聋。其声阻抗检查与纯音电测听检查结果相符。颞骨CT显示内耳结构及发育均无异常。全身检查无其他系统异常,排除内耳及神经的占位病变。第二胎出生后住院期间听力筛查双耳通过,至今已10月龄,随访听力完全正常。
2.2 家系所有成员GJB2基因型测序及比对结果由图3、4可见,测序结果显示父亲携带GJB2 176-191del16杂合突变,母亲携带GJB2 235delC杂合突变。先证者分别遗传了父母有突变的一条染色体,即同时携带235delC与176-191del16的复合突变,因此属于GJB2复合突变引起的常染色体隐性遗传耳聋。因其父母均为携带者,再生育聋儿的风险为25%。胎儿遗传了母亲有突变的第13号染色体,携带GJB2 235delC杂合突变,即一条染色体为GJB2 235delC,而另一条染色体正常,因此,仅为GJB2 235delC杂合突变携带者,发生耳聋的风险与正常人群相近,不同于先证者的基因型,不会复制先证者的听力结构。
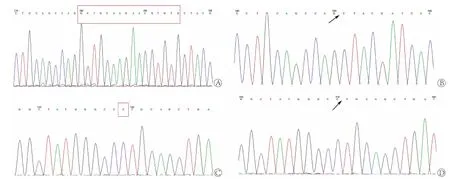
图3 先证者突变型测序图Fig.3 Sequencing results of the mutant of the proband

图4 家系各成员GJB2 DNA序列比对Fig.4 GJB2 DNA sequence alignment of the family
3 讨 论
GJB2突变所致的耳聋主要为先天性NSHI,为常染色体隐性遗传模式。在中国人群中,已检出由GJB2突变所致的儿童语前聋达到26%,占常染色体隐性遗传性耳聋的28%[9-10]。世界不同地区的耳聋家系中均检测到GJB2基因突变[11-17],其中重度耳聋新生儿的发病率达1/800~1/1000[2,18]。近期对不同年龄段NSHI常见基因进行检测发现,GJB2基因突变患者的发病年龄集中在1岁以内,所占比例高达66.67%[19]。由于儿童NSHI无其他组织器官异常表现,因此很难及时筛查和早期干预[20]。本文家系父母均为听觉正常的GJB2突变携带者,先证者同时携带父亲的176-191del16bp和母亲的235delC突变,出生即耳聋并丧失语言能力。胎儿基因检测结果为GJB2基因235delC杂合突变,来源于其母亲,因此胎儿为GJB2 235delC杂合突变携带者,耳聋风险与正常人群接近。后随访证实,该胎儿出生后通过双耳听力筛查,至今已10月龄,听力完全正常。夫妇两人为同一基因(GJB2)致病突变携带者,这类耳聋家庭在临床中所占比例相对较小,但一旦发生,其孕育聋儿的风险将大幅提高。因此,必须注重与普及婚育前耳聋基因普遍性筛查,做好产前诊断及干预。
GJB2基因编码分子量为26.5kD的缝隙连接蛋白26(Connexin26,Cx26),定位于染色体13q11-12,基因组DNA全长4804kb,含有2个外显子、1个内含子,其编码区位于第2外显子[21]。Cx26主要分布于内耳的血管纹、螺旋韧带、螺旋缘及耳蜗的支持细胞之间,与相邻细胞的间隙连接蛋白组成间隙连接通道,维持耳蜗中细胞间信息的正常传递和K+的正常循环,保证耳蜗听觉系统的正常功能[22-24]。GJB2基因发生突变可导致Cx26蛋白结构异常,缝隙连接缺损,通道不能正常开闭,使细胞间信号传递受阻或紊乱,从而引起感音神经性耳聋[21,25]。在我国,GJB2 235delC是NSHI患者的主要突变方式[16,26],对先天性耳聋患者进行突变筛查发现,GJB2 235delC突变在先天性耳聋患者中所占比例为18.16%[27]。235delC突变位于Cx26蛋白第二跨膜区,引起第79位密码子移码突变,导致终止密码子提前至第82位,使所编码的多肽只有81个氨基酸,比野生型蛋白截短了145个氨基酸,造成其间隙连接功能失活,产生了一个无功能的缝隙连接蛋白[28]。176-191del16bp突变是NSHI人群GJB2基因中仅次于235delC突变的另一种突变形式[29],可导致密码子59-76的移码改变,使终止密码子提前,产生无功能蛋白,使K+经内耳毛细胞循环回流进入耳蜗内淋巴液的循环受到影响,最终导致耳聋的发生。该家系在分子水平上验证了携带GJB2基因突变的耳聋为先天性耳聋的特点,提示在临床上进行耳聋患者GJB2基因诊断时,应将235delC和176-191del16突变列为耳聋基因检测的常规项目。
GJB2基因突变所致的耳聋大多数为隐性遗传,其系谱特点为:①患者的双亲表现型往往正常,但他们均为致病基因携带者,出生耳聋患儿的可能性约占1/4,患儿的正常同胞中有2/3的可能性为携带者;②系谱中看不到连续传递,即表现为散发病例,往往在整个系谱中只有先证者1例患者;③性状的遗传与性别无关;④近亲婚配时,后代中发病率明显增高。因此,在产前诊断中,如果发现胎儿携带2个GJB2基因的致病突变位点,则其发生耳聋的概率为100%。我们将家系的分析结果用于先证者家庭的产前筛查与产前诊断,避免了该家庭再次生育突变基因型的儿童。在我国的NSHI患者中,明确为GJB2基因突变引起耳聋的比例达到21.01%[29]。因此对GJB2基因突变导致耳聋的家庭进行遗传咨询、产前筛查和产前诊断显得尤为重要。
耳聋产前诊断应尽可能在孕早期进行,早期得到诊断结果,早期进行生育选择,避免痛苦较大的中后期引产。此外,母亲最好提前进行遗传咨询,在耳聋遗传风险以及致聋基因型确定之后再怀孕,以免造成不良后果[30]。此例家庭的胎儿只携带了来自母亲的235delC杂合突变,未导致听力异常是幸运的,但怀孕前未进行遗传咨询或检查时间较晚则不可取。接受产前诊断时,应根据母亲妊娠的不同时期选择适当的取材方法,妊娠11~13周时行绒毛膜取样,妊娠16~22周时行超声引导下羊膜腔穿刺羊水取样,妊娠22~37周时行超声引导下的脐带穿刺取血(妊娠13~16周时暂不取材)。因此笔者建议,如发现有耳聋先证者,先确定其耳聋病因;在进行了基因突变分析和详尽的遗传咨询后,再决定其家系成员是否受孕和接受产前诊断。只有这样,才能真正达到产前诊断目的,从而有效阻断耳聋在这一高危人群中的传递。
我国每年新生聋儿约3万例,耳聋的基因筛查和产前诊断依然面临艰巨的任务和挑战。随着新一代测序技术的应用,新的耳聋基因将不断被发现和鉴定,这为临床遗传性耳聋的基因诊断提供了新的候选位点,也为遗传咨询和产前诊断提供了新的依据,有助于进一步降低出生缺陷。
[1]Yuan HJ, Cao JY, Sun HJ, et al. Investigation of a huge family with autosomal dominant hereditary non-syndromic hearing loss[J]. Med J Chin PLA, 2003, 28(8): 703-704. [袁慧军, 曹菊阳, 孙捍军, 等. 一个常染色体显性遗传非综合征性耳聋巨大家系调查[J]. 解放军医学杂志, 2003, 28(8): 703-704.]
[2]Wang QJ, Zhao YL, Rao SQ, et al. Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns[J]. Int J Pediatr Otorhinolaryngol,2011, 75(4): 535-542.
[3]Van Camp G, Willems PJ, Smith RJ. Nonsyndromic hearing impairment: unparalleled heterogeneity[J]. Am J Hum Genet,1997, 60(4): 758-764.
[4]Marcolla A, Bouchetemble P, Lerosey Y, et al. Genetic deafness[J]. Ann Otolaryngol Chir Cervicofac, 2006, 123(3):143-147.
[5]Morton CC. Genetics, genomics and gene discovery in the auditory system[J]. Hum Mol Genet, 2002, 11(10): 1229-1240.
[6]Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness[J]. Nature,1997, 387(6628): 80-83.
[7]Walsh T, Shahin H, Elkan-Miller T, et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82[J]. Am J Hum Genet, 2010, 87(1): 90-94.
[8]Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review[J]. Genet Med, 2002, 4(4): 258-274.
[9]Zheng WB, Luo JH, Li Y, et al. Mutations in the GJB2 gene in Chinese patients with prelingual non syndromic hearing impairment[J]. Chin J Pediatr, 2000, 38(10): 610-613.[郑文波,罗建红, 郦云, 等. 中国人语前非综合征性耳聋患者GJB2基因的突变分析[J]. 中华儿科杂志, 2000, 38(10): 610-613.]
[10]Ke XM, Lu Y, Liu YH, et al. Study on mutations in the connexin 26 gene among Chinese with nonsyndromic hearing loss[J].Chin J Otorhinolaryngol, 2001, 36(3): 163-165.[柯肖枚, 路远,刘玉和, 等. Cx26基因突变与国人遗传性无综合征耳聋相关性分析[J]. 中华耳鼻咽喉科杂志. 2001, 36(3): 163-165.]
[11]Morell RJ, Kim HJ, Hood LJ, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness[J]. N Engl J Med, 1998, 339(21): 1500-1505.
[12]Abe S, Usami S, Shinkawa H, et al. Prevalent connexin 26 gene(GJB2) mutations in Japanese[J]. J Med Genet, 2000, 37(1): 41-43.
[13]Sobe T, Vreugde S, Shahin H, et al. The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population[J]. Hum Genet, 2000, 106(1): 50-57.
[14]Gabriel H, Kupsch P, Sudendey J, et al. Mutations in the connexin26/GJB2 gene are the most common event in nonsyndromic hearing loss among the German population[J]. Hum Mutat, 2001, 17(6): 521-522.
[15]Hutchin T, Coy NN, Conlon H, et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK- implications for genetic testing[J]. Clin Genet, 2005, 68(6):506-512.
[16]Dai P, Yu F, Han B, et al. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population[J]. Genet Med, 2007,9(5): 283-289.
[17]Carlsson PI, Karltorp E, Carlsson-Hansen E, et al. GJB2(Connexin 26) gene mutations among hearing-impaired persons in a Swedish cohort[J]. Acta Otolaryngol, 2012, 132(12): 1301-1305.
[18]Steel KP. New interventions in hearing impairment[J]. BMJ,2000, 320(7235): 622-625.
[19]Zhang CQ, Chen BB, Chen YY, et al. Prevalence of common genetic mutations and clinical characteristics analysis in pati ents at different ages with nonsyndromic hearing impairment[J].Hereditas, 2013, 35(3): 352-358. [张初琴, 陈波蓓, 陈迎迎, 等.不同年龄段非综合征性耳聋常见基因检测及临床表型分析[J]. 遗传, 2013, 35(3): 352-358.]
[20]Zhou A, Fang RP, Dai P, et al. Genetic testing of GJB2, SLC26A4 and mtDNA (C494T, A1555G) mutation and prenatal counseling with deafness families of children[J]. J Zhengzhou Univ (Med Sci), 2011, 46(3): 385-388.[周艾, 方如平, 戴朴, 等.儿童耳聋家庭GJB2、SLC26A4和mtDNA基因型检测及产前咨询[J]. 郑州大学学报(医学版), 2011, 46(3): 385-388.]
[21]Forge A, Becker D, Casalotti S, et al. Gap junctions and connexin expression in the inner ear[J]. Novartis Found Symp, 1999, 219:134-150.
[22]Todt I, Hennies HC, Basta D, et al. Vestibular dysfunction of patients with mutations of Connexin 26[J]. Neuroreport, 2005,16(11): 1179-1181.
[23]Kikuchi T, Kimura RS, Paul DL, et al. Gap junctions in the rat cochlea: immunohitochemical and ultrastructural analysis[J].Anat Embryol, 1995, 191(2): 101-118.
[24]Holt JR, Corey DP. Ion channel defects in hereditary hearing loss[J]. Neuron, 1999, 22(2): 217-219.
[25]Lefebvre PP, Van De Water TR. Connexins, hearing and deafness: clinical aspects of mutations in the connexin 26 gene[J]. Brain Res Brain Res Rev, 2000, 32(1): 159-162.
[26]Xu YF, Ren LF, Song WQ , et al. Mutation analysis of Cx26 gene in Chinese hereditary nonsyndromic deafness sufferers[J]. Chin J Otorhinolaryngol, 2002, 37(5): 348-351. [徐悦凡, 任鲁风, 宋文芹, 等. 中国人非综合征型听力损失患者Cx26基因的突变分析[J]. 中华耳鼻咽喉科杂志, 2002, 37(5): 348-351.]
[27]Dai P, Liu X, Yu F, et al. Molecular etiology of patients with nonsyndromic hearing loss from deaf-mute schools in 18 provinces of China[J]. Chin J Otol, 2006, 4(1): 1-5. [戴朴, 刘新, 于飞, 等. 18个省市聋校学生非综合征性聋病分子流行病学研究(I)-CJB2 235delc和线粒体 DNA 12S rRNA A1555G突变筛查报告[J]. 中华耳科学杂志, 2006, 4(1): 1-5.]
[28]Choung YH, Moon SK, Park HJ. Functional study of GJB2 in hereditary hearing loss[J]. Laryngoscope, 2002, 112(9): 1667-1671.
[29]Yu F, Han DY, Dai P, et al. Mutation of GJB2 gene in nonsyndromic hearing impairment patients: analysis of 1190 cases[J]. Natl Med J Chin, 2007, 87(40): 2814-2819. [于飞, 韩东一, 戴朴, 等. 1190例非综合征性耳聋患者GJB2基因突变序列分析[J]. 中华医学杂志, 2007, 87(40): 2814-2819.]
[30]Hu H, Wu LQ, Liang DS, et al. Prenatal diagnosis of pre lingual deafness by determ ination of SLC26A4 genemutation[J]. Chin J Obstet Gynecol, 2005, 40(9): 591-594. [胡浩, 邬玲仟, 梁德生, 等. 学语前性耳聋疾病相关基因的产前诊断[J]. 中华妇产科杂志, 2005, 40(9): 591-594.]
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
中国民间疗法(2021年8期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
中国生殖健康(2018年4期)2018-11-06
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
重庆医学(2015年12期)2015-03-05
