
人参皂苷M1异丁酸酯衍生物的合成及高效液相分析和制备△
2014-11-03 00:41肖景楠李珂珂李争宁陈丽荣弓晓杰
中国现代中药 2014年5期
肖景楠,李珂珂,李争宁,陈丽荣,弓晓杰*
(1.大连大学 环境与化学工程学院,辽宁 大连 116622;2.大连大学 医学院,辽宁 大连 116622)
研究开发
国家自然科学基金(81172949);辽宁省教育厅科学技术研究一般研究项目(L2012439);辽宁省优秀人才支持计划(LR2013058);辽宁省科技厅科学技术计划项目(2013204001);大连市科技局科技计划项目(2013E11SF055)
*
弓晓杰,男,教授,硕士生导师,研究方向:中药新药开发;E-mail:gxjclr@163.com
人参皂苷M1异丁酸酯衍生物的合成及高效液相分析和制备△
肖景楠1,李珂珂2,李争宁1,陈丽荣1,弓晓杰2*
(1.大连大学 环境与化学工程学院,辽宁 大连 116622;2.大连大学 医学院,辽宁 大连 116622)
目的研究人参皂苷M1异丁酸酯合成过程中产物的高效液相分析及快速制备方法。方法采用保护-酯化-去保护的方法,定向修饰得到M1糖基3-位酯化的产物;利用反相高效液相色谱,采用DAD检测器,通过对分析条件的优化,建立人参皂苷M1异丁酸酯及其两个中间产物的分析制备方法。结果人参皂苷M1异丁酸酯及其中间产物的分析和制备时间均在40 min以内快速高效地获得;综合三步反应和高效液相制备结果,合成人参皂苷M1异丁酸酯的得率为36.7%,纯度为98.3%。结论采用HPLC法,在M1衍生物合成过程中可以快速高效地进行人参皂苷M1衍生物的分析和制备。
人参皂苷M1;结构修饰;高效液相;含量分析
人参皂苷是人参的主要活性成分,目前人参皂苷在动物体内的代谢已成为一个重要的研究方向,通过体内代谢产物的分析来明确人参皂苷实际发挥药效的成分[1-2]。大量的研究表明,口服人参后被肠道菌群代谢得到的人参皂苷M1是体内的主要代谢产物,并且M1又进一步在肝脏中与脂肪酸酯化转化为EM1[3-5],从而延长其在体内的存留时间,表现出更强的活性作用,如抗肿瘤活性[6]。在已完成的实验研究中,我们合成了不同碳链长度、糖基上不同取代位置的多种人参皂苷M1脂肪酸酯衍生物,并研究了其抗肿瘤活性和构效关系[7-8]。
在本研究中,我们采用保护-酯化-去保护的策略,有选择性地合成3-位糖基酯化产物,并利用反相高效液相色谱方法,建立了中间产物、终产物的分析和制备方法,为快速、大量地分析和制备人参皂苷修饰物奠定了理论和实验基础。
1 仪器、试剂和材料
1.1 仪器
高效液相色谱仪:[分析型Agilent 1260,Agilent C18色谱柱(250 mm×4.6 mm,5 μm);制备型LC 3000,C18色谱柱(250 mm×10 mm,5 μm),电子天平(上海梅特勒-托利多仪器有限公司),ETS-D4加热磁力搅拌器(德国IKA公司)。
1.2 试剂
甲醇为色谱纯,水为超纯水,其他试剂均为分析纯。
1.3 材料
人参皂苷M1(大连富生天然药物有限公司,批号:20100321),经NMR及MS测定纯度为98%。人参皂苷M1的硅醚衍生物、人参皂苷M1硅醚衍生物的异丁酸酯及人参皂苷M1的异丁酸酯,自制并鉴定结构。
2 方法和结果
2.1 衍生物的合成
2.1.1 人参皂苷M1叔丁基二甲基硅醚的合成 人参皂苷M1与5倍量的叔丁基二甲基氯硅烷在氮气保护下,以三乙胺为溶剂在室温下反应24 h后,加入饱和氯化铵溶液终止反应。经二氯甲烷(CH2Cl2)萃取、Brine洗有机相、无水Na2SO4干燥并浓缩溶剂[9]。
2.1.2 人参皂苷M1叔丁基二甲基硅醚异丁酸酯的合成 将第一步纯化后的人参皂苷M1的叔丁基二甲基硅醚与5%当量的DMAP在氮气保护、冰水浴下加入适量的CH2Cl2溶解,与三乙胺反应10 min后,加入等当量的异丁酰氯,反应24 h,然后加入饱和氯化铵溶液停止反应。经CH2Cl2萃取,Brine洗有机相,无水Na2SO4干燥,浓缩溶剂[7]。
2.1.3 人参皂苷M1异丁酸酯的合成 将液相制备的人参皂苷M1硅醚衍生物的异丁酸酯加入到50 mL的圆底烧瓶中,用无水四氢呋喃溶解,加入1.5倍量的四丁基氟化铵,反应2 h后停止。旋干无水四氢呋喃溶液,加入CH2Cl2溶解并用其萃取,Brine洗有机相,无水Na2SO4干燥,浓缩溶剂[10-11]。合成反应如图1所示。
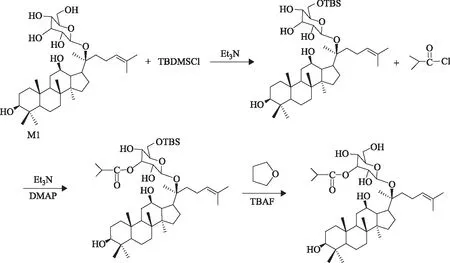
图1 人参皂苷M1异丁酸酯的合成路线
2.2 高效液相分析条件的确定
2.2.1 色谱柱的选择 分别采用Agilent C18不同长度及型号的色谱柱进行合成产物的分离实验。综合实验结果,本实验选用Zorbax Eclipse XDB-C18(250 mm×4.6 mm,5 μm)色谱柱,可以达到较好的分离效果。
2.2.2 检测波长的选择 人参皂苷M1的单醚化衍生物采用DAD检测器进行190~400 nm检测时,发现其单醚化衍生物在203 nm下有最大吸收[12]。因而在实验操作中采用203 nm作为检测波长。
2.2.3 流动相的选择 实验中根据人参皂苷M1合成衍生物的结构特征,在乙腈-水体系和甲醇-水体系之间进行了筛选。从分离度和峰形来看,95%甲醇-水体系较适合作为分离人参皂苷M1单醚化衍生物的流动相。
2.2.4 流速的选择 本实验参照文献报道[13]对流动相的流速也进行了条件优化,采用甲醇-水体系为流动相,在流速为1.0 mL·min-1时,合成产物中杂质峰与目标峰分离效果较好,且目标峰的出峰时间适宜,故流速定为1.0 mL·min-1。
2.2.5 柱温的选择 考查了不同温度下的衍生物分离度,实验结果发现,柱温为25 ℃时,分离效果较好。
2.3 中间产物及终产物高效液相分析和制备
2.3.1 人参皂苷M1叔丁基二甲基硅醚的测定 将合成产物制备成浓度为5.0 mg·mL-1的甲醇溶液,经0.45 μm微孔滤膜过滤,采用2.2的色谱条件,进样10 μL。得到的人参皂苷M1叔丁基二甲基硅醚的色谱图见图2。
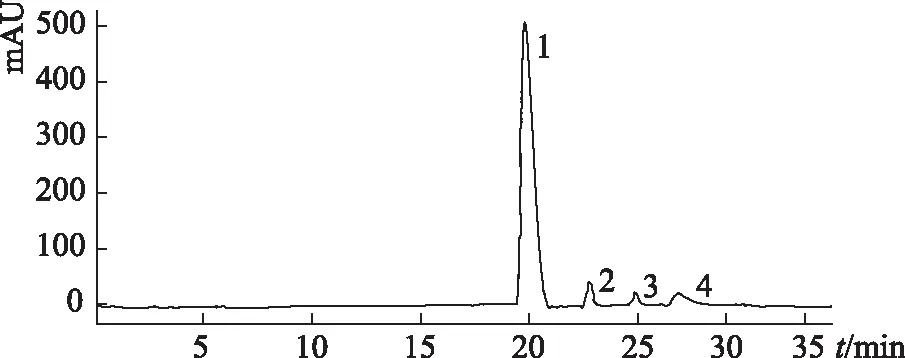
图2 人参皂苷M1叔丁基二甲基硅醚的液相色谱图
从色谱图中可以看出,连接上保护基后产物有4个,其中1号峰为人参皂苷M1叔丁基二甲基硅醚。根据峰面积百分比计算法,其占产物含量的84.06%,见表1。
2.3.2 人参皂苷M1叔丁基二甲基硅醚的制备 采用分析型高效液相色谱建立的色谱条件,经制备型高效液相纯化,得到的色谱图见图3。其中37.14 min(峰1)所出的色谱峰即为人参皂苷M1叔丁基二甲基硅醚。收集该处色谱峰流出液、浓缩,即得第一步反应产物。

表1 人参皂苷M1叔丁基二甲基硅醚合成产物中化合物出峰时间及相应峰面积百分比

图3 人参皂苷M1叔丁基二甲基硅醚的制备液相色谱图
2.3.3 人参皂苷M1叔丁基二甲基硅醚的异丁酸酯的测定 将合成产物制备成浓度为5.0 mg·mL-1的甲醇溶液,经0.45 μm微孔滤膜过滤,采用2.2的色谱条件,进样10 μL。得到的人参皂苷M1叔丁基二甲基硅醚的异丁酸酯的色谱图见图4。
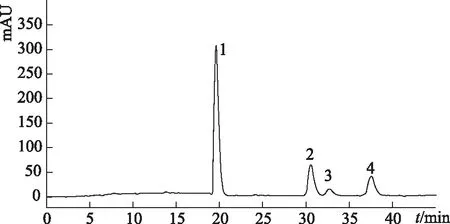
图4 人参皂苷M1叔丁基二甲基硅醚与异丁酰氯酯化产物的液相色谱图
从色谱图中可以看出,酯化后的产物有4个,其中1号峰为酯化产物。根据峰面积百分比计算法,其占产物含量的77.59%,见表2。
2.3.4 人参皂苷M1叔丁基二甲基硅醚的异丁酸酯的制备 采用分析型高效液相色谱建立的色谱条件,经制备型高效液相纯化,得到的色谱图见图5。其中36.95 min所出的色谱峰(峰1)即为人参皂苷M1叔丁基二甲基硅醚的异丁酸酯。收集该处色谱峰流出液,浓缩,即得第二步反应产物。

表2 人参皂苷M1叔丁基二甲基硅醚异丁酸酯合成产物的出峰时间及相应峰面积百分比
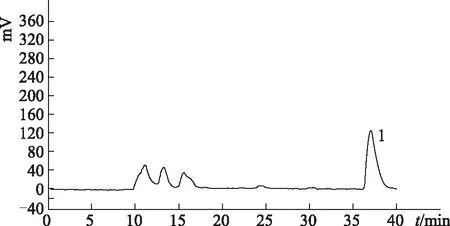
图5 人参皂苷M1叔丁基二甲基硅醚与异丁酰氯酯化产物的制备液相色谱图
2.3.5 人参皂苷M1异丁酸酯的测定 将脱去硅醚保护基的合成产物制备成浓度为5.0 mg·mL-1的甲醇溶液,经0.45 μm微孔滤膜过滤,采用2.2的色谱条件,进样10 μL。得到的人参皂苷M1异丁酸酯的色谱图见图6。

图6 人参皂苷M1异丁酸酯的液相色谱图
从色谱图中可以看出,脱保护基后的产物有3个,其中1号峰为目标产物。根据峰面积百分比计算法,其占产物含量的58.65%,见表3。

表3 人参皂苷M1异丁酸酯合成产物的出峰时间及相应峰面积百分比
2.3.6 人参皂苷M1异丁酸酯的制备 采用分析型高效液相色谱建立的色谱条件,经制备型高效液相纯化,得到的色谱图见图7。其中34.59 min(峰1)所出的色谱峰即为人参皂苷M1异丁酸酯。收集该处色谱峰流出液、浓缩,即得反应终产物。
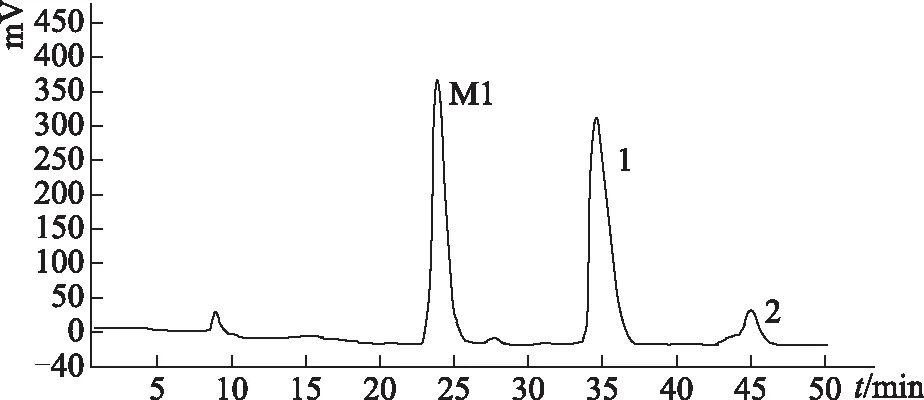
图7 人参皂苷M1异丁酸酯的制备液相色谱图
2.4 人参皂苷M1异丁酸酯得率的计算
投入人参皂苷M1 100.0 mg,液相制备后得到纯人参皂苷M1叔丁基二甲基硅醚(M1-OTBS)98.4 mg,可以得出第一步反应的产率为83.2%;第二步酯化反应投入M1-OTBS总量为95.0 mg,液相制备后得到纯的目标产物为77.8 mg,酯化产率为74.8%;最后脱去硅醚保护基的反应投入人参皂苷M1硅醚的异丁酸酯为75.0 mg,液相制备后得到纯的人参皂苷M1异丁酸酯为37.5 mg,脱保护反应产率为58.3%。由以上三步反应的产率可以得到人参皂苷M1经过保护-酯化-去保护策略生成人参皂苷M1异丁酸酯的得率为36.7%。
3 讨论
3.1 人参皂苷M1叔丁基二甲基硅醚的液相结果分析
由于人参皂苷M1的糖苷基上的6位羟基最为活泼,所以人参皂苷M1与叔丁基二甲基氯硅烷反应的主产物为糖苷基6位上的单硅醚,这个过程伴随生成多种人参皂苷M1硅醚异构体,这些副产物与糖苷基上6位硅醚的人参皂苷M1衍生物极性相似,难以分离提纯,而我们在高效制备液相上可以很理想地将其分开,液相色谱图(见图2)上主产物的出峰时间为19.89 min,整合合成人参皂苷M1的各硅醚衍生物的峰面积,可以得出糖苷基6位上人参皂苷M1单硅醚衍生物在醚化后样品内的含量为84.06%,并将优化后的液相条件应用于人参皂苷M1硅醚衍生物的液相制备。在制备过程中,未把2~4号峰全部分流收集,将其混入下一针的制备中流入废液以节省时间、节约试剂。从结果来看,不影响人参皂苷M1叔丁基二甲基硅醚的纯度,收集物纯度为97.5%。
3.2 人参皂苷M1硅醚衍生物与异丁酰氯酯化产物的液相结果分析
人参皂苷M1的硅醚衍生物与等当量的异丁酰氯酯化过程,根据位阻大小的影响,反应的主产物为糖苷基3位上异丁酸酯的人参皂苷M1衍生物[14-15],其出峰时间为19.61 min,其在酯化反应后样品内的含量为77.59%,通过液相制备后纯度达到96.8%。在制备过程中,图4中的2~4号峰亦未收集流分,转入下一针的废液中,即制备色谱图(图5)中11.14、13.26、15.58 min的3个峰。
3.3 人参皂苷M1衍生物脱去硅烷保护基后的产物液相结果分析
人参皂苷M1衍生物脱去硅烷保护基的同时有部分酯基发生水解生成了人参皂苷M1,人参皂苷M1异丁酸酯的出峰时间为11.26 min,在脱去保护基后的样品内含量为58.65%,液相制备后人参皂苷M1异丁酸酯的纯度为98.3%。
综上,本文通过高效液相色谱法准确分析得到人参皂苷M1合成衍生物中目标产物的含量,探索出最佳的人参皂苷M1合成衍生物的液相制备条件,包括检测波长、流动相组成、流速等,确定了样品中各组分出峰时间,并将优化后的条件应用于液相制备,从而达到快速准确分离的目的,解决了人参皂苷M1合成目标衍生物与反应过程中伴随生成的异构体之间所存在的分离提纯难题,产物损失量小且方法简捷可行。
[1] Lee J,Lee E,Kim D,et al.Studies on absorption,distribution and metabolism of ginseng in humans after oral administration[J].J Ethnopharmacol,2009,122(1): 143-148.
[2] Qian TX,Cai ZW.Biotransformation of ginsenosides Rb1,Rg3and Rh2in rat gastrointestinal tracts[J].Chin Med,2010,5:19-25.
[3] Hasegawa H,Lee KS,Nagaoka T,et al.Pharmacokinetics of ginsenoside deglycosylated by intestinal bacteria and its transformation to biologically to biologically active fatty acid esters[J].Biol Pharm Bull,2000,23(3): 298-304.
[4] Bae EA,Choo M,Park EK,et al.Metabolism of ginsenoside Rc by human intestinal bacteria and its related antiallergic activity[J].Biol Pharm Bull,2002,25(6):743-747.
[5] Tawab MA,Bahr U,Karas M,et al.Degradtion of ginsenosides in humans after oral administration[J].Drug Metab Dispos,2003,31(8):1065-1071.
[6] Bae EA,Han MJ,Choo MK,et al.Metabolism of 20(S)-and 20(R)-ginsenoside Rg3by human intestinal bacteria and its relation to in vitro biological activities[J].Biol Pharm Bull,2002,25(1):58-63.
[7] Li WF,Li ZN,Chen LR,et al.Synthesis and structural analysis of mono-dodecanoic acid esters of ginsenoside M1[J].Chin J Nat Med,2011,9(3):199-203.
[8] Li WF,Chen LR,Gong XJ,et al.Synthesis of esters of ginsenoside metabolite M1 and their cytotoxicity on MGC80-3 cells[J].Molecules,2013,18(4):3689-3702.
[9] Kawabata T,Muramatsu W,Nishio T,et al.A catalytic one-step process for the chemo-and regioselective acylation of monosaccharides[J].J Am Chem Soc,2007,129(42):12890-12895.
[10] White JD,Amedio Jr JC,Gut S,et al.Synthesis of the macrolactone alkaloid(+)-usaramine via necic acid coupling to a pyrrolizidine borane[J].J Org Chem,1989,54(18):4268-4270.
[11] Chen HL,Huang YM,Chang CT,et al.Dicarboxylic acid ester derivatives of ginsenoside,pharmacevical preparations containing the same,and preparation thereof: United States Patent,WO2006113495 A2[P].2006-10-26.
[12] 郑长龙,鱼红闪,金凤燮.人参稀有皂苷C-K的分离与纯化[J].大连工业大学学报,2003,22(3):167-169.
[13] 周伟,罗振时.反相高效液相色谱法测定人参皂苷Compound-K的含量[J].色谱,2005,23(3):270-272.
[14] 孙印石,刘政波,孙彦君,等.RP-HPLC法测定人参皂苷Compound K 的合成产物棕榈酸酯PM1的转化率[J].天然产物研发与开发,2007,19(2):309-312.
[15] Jansson PE,Kenne L,Schweda E.Nuclear magnetic resonance and conformational studies on monoacetylated methyl D-gluco-and D-galacto-pyranosides[J].J Chem Soc Perkin Trans,1987,1: 377-383.
SynthesisofGinsenosideM1IsobutyrateanditsAnalysisandPreparationbyHPLC
XIAOJingnan1,LIKeke2,LIZhengning1,CHENLirong1,GONGXiaojie2
(1.CollegeofEnvironmentalandChemicalEngineering,DalianUniversity,Dalian116622,China;2.CollegeofMedical,DalianUniversity,Dalian116622,China)
Objective:Ginsenoside M1 isobutyrate and the intermediate product in the process of synthesis were analyzed and prepared by HPLC.MethodsUsing the protection esterification-deprotection strategy,ginsenoside M1 3-sugar residues esterification product was synthesed.The analysis and preparation method of M1 isobutyrate and the intermediate product by RP-HPLC-DAD was established and optimized.ResultsThe analysis time of ginsenoside M1 isobutyrate and the intermediate product was all less than 40 minutes.Taking all the three steps of synthesis into account,the yield was 36.7%,and purity was 98.3%.ConclusionThe HPLC method put forward a rapid and efficient strategy to deal with the products through the process of ginsenoside M1 synthesis.
Ginsenoside M1;Structural modification;HPLC;Content analysis
10.13313/j.issn.1673-4890.2014.05.005
2014-04-01)
猜你喜欢
当代水产(2022年1期)2022-04-26
当代水产(2022年2期)2022-04-26
太原理工大学学报(2022年2期)2022-03-21
中国饲料(2021年17期)2021-11-02
化工环保(2021年3期)2021-06-17
当代水产(2021年2期)2021-03-29
上海计量测试(2020年1期)2020-03-18
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
百科知识(2016年18期)2016-10-28
