
叶酸偶联载多西紫杉醇壳聚糖纳米粒包封率的测定
2014-11-02 04:00汪小乐邓燕芬庞廷媛程国华
现代医院 2014年2期
汪小乐 邓燕芬 庞廷媛 程国华
多西紫杉醇(DTX)是一种半合成的紫杉烷类抗肿瘤药物,抗癌活性比紫杉醇较高。其作用机制是增强微管蛋白聚合并抑制微管解聚,导致形成稳定的非功能性维管束,破坏有丝分裂和细胞增殖,是现有药物中治疗转移乳腺癌和非小细胞肺癌最有效的单剂化疗药物[1]。目前国内上市的剂型为注射液,因处方中含有大量非离子表面活性剂吐温-80和增溶剂无水乙醇,常常导致临床严重的过敏反应。同时,由于药物的分布无选择性,易导致中性粒细胞减少、神经毒性等毒副作用[2]。为降低其毒副作用,提高制剂的肿瘤靶向性和临床疗效,本试验将DTX制备成叶酸偶联载多西紫杉醇壳聚糖纳米粒,并采用离心法分离载药纳米粒中的游离药物,以高效液相色谱法测定DTX的含量及包封率,并进行方法学考察。
1 仪器与材料
BT125D Sartorius电子天平(赛多利斯天平有限公司);RW20DZM.n强力流线型搅拌机(IKA,德国);LC-20AT型高效液相色谱仪配SPD-20A型检测器;LCsolution色谱工作站;CENTRIFUGE5424离心机(eppndorf)。
壳聚糖(DAC degree≥ 95%,分子量161.16 KDa);多西紫杉醇(DTX,江苏恒瑞医药股份有限公司,批号:626111003,含量101.0%);多西紫杉醇对照品(恒瑞医药股份有限公司,批号:26070904,含量99.0%);N-羟基丁二酰亚胺(NHS,成都市科龙化工试剂厂,批号:20110201);N,N'-二环己基碳二亚胺(DCC,阿拉丁,批号:48623);叶酸(FA,科龙化工试剂厂,批号:20101001,含量≥97.0%);无水二甲基亚砜(DMSO);其余试剂均未分析纯,超纯水为实验室自制。
2 方法与结果
2.1 FA-CTS/DTX纳米粒及空白FA-CTS纳米粒的制备
2.1.1 叶酸活性酯的制备 称取FA 0.3 g、DCC 0.282 g、NHS 0.156 g,溶于10 ml无水DMSO中,室温避光反应过夜,过滤除去反应副产物二环己基脲,滤液在搅拌下中逐滴滴入冰冷的含30%丙酮的无水乙醚溶液中,得黄色沉淀物,再用无水乙醚洗2遍,真空干燥,得叶酸活性酯。
2.1.2 叶酸偶联壳聚糖的制备 称取壳聚糖20 mg,溶于5 ml醋酸-醋酸钠缓冲液中,磁力搅拌下缓慢加入1 ml叶酸活性酯的DMSO溶液(20 mg/ml),30℃下避光反应16 h后,调节溶液pH至9.0。将沉淀出的叶酸偶联壳聚糖用蒸馏水洗涤数遍后,重新溶于1%醋酸溶液,然后通过SepHadex G-10凝胶柱进行分离,除去尚未反应的游离叶酸。洗脱液为1%醋酸溶液,流速1.5 ml/min。运用紫外可见分光光度计在363 nm处监测洗脱过程,收集第一个流出的峰,冷冻干燥后备用[3]。
2.1.3 FA-CTS/DTX纳米粒的制备 将FA-CTS冻干粉末溶于1%醋酸溶液中得3 mg/ml FA-CTS溶液。称取0.5 mg DTX粉末溶于适量无水乙醇中,超声至完全溶解,形成多西他赛无水乙醇溶液。将两者混合,持续磁力搅拌下,缓慢加入2 mg/ml STPP溶液,磁力搅拌30 min,得到FA-CTS/DTX纳米粒。按同样反应条件,制备不加DTX的空白FA-CTS纳米粒。
2.2 含量测定
2.2.1 色谱条件 色谱柱:Hypersil ODS2(250 mm×4.6 mm,5 μm);流动相:甲醇 - 水(67∶33);流速:1.0 ml/min;柱温:40 ℃;检测波长:229 nm;进样量:10 μl。
2.2.2 标准曲线绘制 精密称取DTX对照品5.0 mg,加入无水乙醇溶解定容至10 ml量瓶,得浓度为0.5 mg/ml的DTX储备液。精密移取该储备液,配置成0.1、0.5、1.0、1.5、15.0 μg/ml的对照品溶液。分别取上述对照液10 μl进样,以峰面积(Y)为纵坐标,以DTX质量浓度(X)为横坐标进行线性回归分析,得回归方程:Y=26 392X+1 147.5,r=0.999 9。多西紫杉醇浓度在 0.1 ~15.0 μg/ml范围内呈良好的线性关系。
2.2.3 精密度试验 取2.2.2项下0.1、1.0、15.0 μg/ml三种质量浓度对照液分别重复进样6次,RSD分别为1.67%、0.14%、0.12%。表明本方法精密度良好。
2.2.4 回收率试验 取 2.2.2项下 DTX储备液(0.5 mg/ml),分别精密移取 0.8、1.0、1.2 ml置于 10 ml容量瓶中,各加入空白FA-CTS纳米粒1 ml,用无水乙醇定容至10 ml,摇匀,超声后,5 000 r/min离心3 min,取上清液过0.45 μm滤膜后进样分析,按外标法计算主药含量,并计算回收率,结果见表 1。总平均回收率为97.73%,RSD为0.71%。

表1 回收率试验结果
2.2.5 样品含量测定 精密量取FA-CTS/DTX纳米粒1.0 ml,加无水乙醇定容于10 ml容量瓶,摇匀,超声后,5 000 r/min离心3 min,取上清液过0.45 μm滤膜,分别平行制备3分。分别取10 μl进样分析,按外标法计算DTX含量。测得样品平均含量为0.496 5 mg。
2.3 离心法测定包封率
2.3.1 回收率试验 精密吸取FA-CTS/DTX纳米粒样品0.4、0.45和0.475 ml于3个1.5 ml EP管中,加入等体积的DTX储备液,稀释至1.5 ml,混匀,得终浓度分别为0.26、0.30和0.32 mg/ml,分别平行制备3份。移取500 μl上述混合溶液分别于3 000 r/min离心10 min,再分别移取等量上清液至10 ml容量瓶中,稀释至刻度线,摇匀;沉淀也依次用稀释定容至10 ml容量瓶,摇匀,分别测得上清液和沉淀的主药含量,即得0.26、0.30和0.32 mg/ml三个浓度离心后的主药含量。移取500 μl上述混合溶液至10 ml容量瓶中,稀释至刻度线,摇匀,即得3个浓度未经离心处理的总药溶液。按2.2.1项色谱条件测定,按外标法计算主药含量,并计算其回收率。3种浓度的平均回收率分别为101.48%、100.89%和100.92%,总体平均回收率为101.10%,RSD为2.06%。见表2。
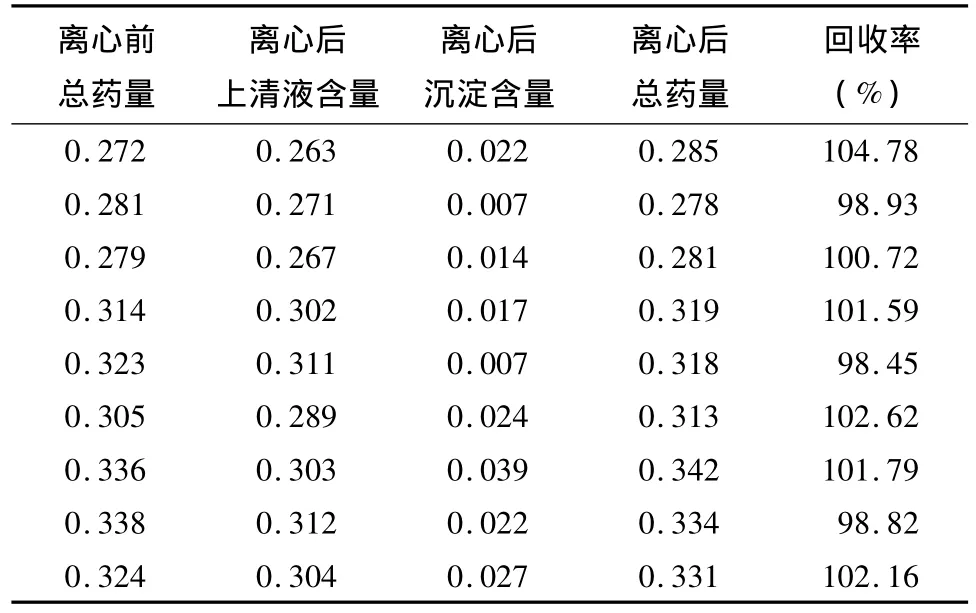
表2 FA-CTS/DTX纳米粒回收率 (mg/ml)
2.3.2 重复性实验 精密吸取FA-CTS/DTX纳米粒样品适量于6个1.5 ml EP管中,加入一定量的DTX储备液,充分混匀,得终浓度为0.30 mg/ml,分别于 3 000 r/min离心10 min,再分别移取等量上清液至10 ml容量瓶,稀释至刻度线,摇匀,按2.2.1项色谱条件测定,按外标法计算主药含量,即得被包封药物含量;同时制备未经离心处理的样品溶液,按2.2.1项色谱条件测定,按外标法计算主药含量,即得总药含量。结果FA-CTS/DTX纳米粒平均包封率为83.74%,RSD为1.3%,离心法测定包封率的重复性良好。
2.3.3 包封率的测定 精密移取适量FA-CTS/DTX纳米粒于1.5 ml EP管中,3 000 r/min离心10 min,精密吸取上清液及未经离心处理的纳米粒溶液适量,按含量测定方法分别测定其含量,即得包封药物含量(W包)和纳米粒总药含量(W总),按公式计算包封率,包封率=(W包/W总)×100%。3批样品的包封率分别为86.12%、85.94%和85.16%。
3 讨论
包封率是评价纳米粒制备工艺的重要指标,也是纳米粒能否发挥较普通制剂高效、低毒特点并提高药物治疗效果、降低药物毒副作用并减少药物剂量的关键。纳米粒包封率测定的常用方法有葡聚糖凝胶过滤法、超速离心法、微柱离心法[4]、透析法及超滤法等。笔者曾尝试用葡聚糖凝胶过滤法和透析法分离FA-CTS/DTX纳米粒中的游离药物,结果表明凝胶过滤法的分离效果与纳米粒粒径有关,粒径大的粒子柱内通路较短,色谱保留性能较弱,载药纳米粒与游离药物的分离度不好,样品回收率和包封率较低[5]。而多西紫杉醇为脂溶性药物,其在水中溶解度较小,需要较长的透析时间和很大的透析液体,导致游离的药物难以定量检测。
采用低速离心法[6]分离纳米粒和游离药物,具有操作简便、仪器要求低等优点。该方法的关键在于离心强度选择,离心强度过小导致药物沉降不完全,过大则会将载药纳米粒沉淀下来,难以达到分离目的。多西紫杉醇不溶于水,游离的多西紫杉醇大部分以晶体形式析出,本试验采用3 000 r/min离心10 min的条件可将载药纳米粒与游离药物有效分离,方法学验证表明该方法可行,测得FACTS/DTX纳米粒的平均包封率为85.74%。
[1]梁晓丽,夏路风,刘凤琴.多西紫杉醇的药理与临床应用[J].首都医药,1999(5):28-29.
[2]饶 媚,郭丽珍,赖剑锋,赖小平.含紫杉醇联合方案引起肝功能异常的临床分析[J].现代医院,2013,13(10):5 -7.
[3]WARD C M,ACHESON N,SEYMOUR L W.Folic acid targeting of protein conjugates into ascites tumour cells from ovarian cancer patients[J].J Drug Target,2000,8(2):119 - 123.
[4]崔 腾,郭伟英.微柱离心-HPLC法测定卡培他滨脂质体包封率[J].中国医药工业杂志,2012(8):682 -684.
[5]赵 锋,栾瀚森,罗华菲,等.葡聚糖凝胶色谱法用于纳米粒包封率的测定[J].中国药学杂志,2012(17):1385-1390.
[6]王永利,王立华,夏明华,等.低速离心法测定大黄酚脂质体包封率[J].河北北方学院学报:自然科学版,2012(6):30-32,35.
猜你喜欢
中国应急管理科学(2021年4期)2021-04-13
中国生殖健康(2020年6期)2020-02-01
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
中国美容医学(2019年1期)2019-03-01
中国生殖健康(2019年12期)2019-01-07
中国生殖健康(2018年6期)2018-11-06
考试周刊(2018年68期)2018-09-17
中国测试(2018年4期)2018-05-14
妈妈宝宝(2017年4期)2017-02-25
