
第一性原理计算钛酸铅A位掺杂对其负热膨胀性的影响
2014-10-18 05:28:16王方方曹战民邢献然
物理化学学报 2014年8期
王方方 曹战民 陈 骏 邢献然,*
(1北京科技大学冶金与生态工程学院,物理化学系,北京 100083;2北京科技大学冶金与生态工程学院,有色金属冶金系,北京 100083)
1 引言
钛酸铅是一种典型的钙钛矿结构铁电体,在介电、压电、铁电、热电、释电、热膨胀等方面具有重要的理论研究与应用价值.2003年,Xing等1对PbTiO3的单胞参数随温度的变化作了详细的研究,表明其在室温至490°C范围内呈现负热膨胀性,平均本征热膨胀系数αV=-1.99×10-5°C-1,490 °C以上呈现正膨胀性,αV=3.55×10-5°C-1.若以负热膨胀(NTE)材料钛酸铅为基,通过加杂或固溶方式合成ABO3化合物,实现热膨胀系数调控,就可以对钛酸铅基功能陶瓷的性能进行设计和剪裁.2,3钛酸铅的奇异性在于从室温到居里温度,热膨胀系数为负,即随着温度升高体积收缩,这类材料统称为负热膨胀材料.4目前,负热膨胀材料在基础研究和技术应用上面广受关注,5-7这类材料主要有金属氰化物、因瓦合金、纳米晶体等.8-11不同的化合物也拥有着不同的负热膨胀机理:BiNiO3的负热膨胀性产生的原因是金属间的电子转移,12ZrW2O8则是因为刚性模型等等.13近十年来,本实验室利用实验方法对PbTiO3基化合物的负热膨胀性进行了系统的研究,发现其负热膨胀系数与其室温下轴比关系密切,PbTiO3基化合物的晶体结构精细修正结果和拉曼光谱晶格动力学研究表明,纵向光学振动模与横向光学振动模(LO-TO)的劈裂程度和Pb―O(II)的键长、晶胞畸变程度以及负热膨胀变化趋势具有高度一致性,14但其负膨胀的本质仍处于探索过程中.目前所研究的A位掺杂体系中,大部分原子掺杂都会使得钛酸铅的负热膨胀程度有所降低,如Pb1-xSrxTiO3,Pb1-xBixTiO3,Pb1-xLaxTiO3,Pb1-x(La0.5K0.5)xTiO3.15而Cd原子掺杂却使得钛酸铅负热膨胀性增强,热膨胀系数达到-2.40×10-5°C-1.16为了揭示上述现象的原因,我们计算了Cd掺杂钛酸铅的化学键,17发现Cd的掺杂增大了A位原子与氧的共价性,显然进一步比较Cd原子与其他二价原子在掺杂钛酸铅时化学键的异同,将有助于阐明共价性和负热膨胀性的关系,进而深刻地理解钛酸铅负热膨胀的本质.
近年来,随着算法和计算机硬件的改进,量子力学方法更适用于实际材料体系.18第一性原理的优势在于几乎不采用带有经验的参数,而是直接从给定的原子构型开始用量子力学方法进行基本计算,因而具有很高的准确度和预见性.目前利用第一性原理对PbTiO3基化合物的理论研究涉及铁电性、稳定性、热振动、压电性等.19-22Cohen23通过第一性原理计算指出,Ti 3d轨道与O 2p轨道杂化是铁电性的起源.同时,Pb 6s轨道与O 2p轨道也存在共振特性,它们的共同作用使得钛酸铅在室温下能够保持很高的晶格畸变(轴比c/a=1.064).电子结构的第一性原理计算在讨论钛酸铅基化合物物理性质上面也起到了至关重要的作用.24比如,可以将第一性原理与最大熵法结合,研究PbTiO3-BiFeO3化合物的电子结构和电荷密度,结果表明(Bi,Pb)(6s,6 p)轨道与O 2p轨道的杂化是导致化合物具有极大四方性的原因.25综上所述,第一性原理可以研究物质的化学键,这为研究钛酸铅的负热膨胀提供了便利条件.本文采用密度泛函理论研究了A位掺杂钛酸铅的晶格结构和电子结构,比较不同的原子掺入钛酸铅A位后发生的化学键变化,结合实验上对热膨胀系数的测试结果,总结不同A位掺杂钛酸铅的热膨胀性与原子杂化情况的关系.
2 计算方法
利用基于密度泛函理论的第一性原理方法,使用VASP软件包对钛酸铅A位掺杂Sr原子,Ba原子的晶体结构、态密度、电子结构等进行了计算,并与之前已经计算过的Cd原子各物理性质进行比较.交换关联泛函使用了广义梯度近似(GGA),26并选取了PBE对交换关联势的表述,27平面波截断动能选取在400 eV,布里渊区的积分采用了Monkhorst-Pack特殊k点取样方法,k网格点为2×2×2.我们先建立了3×3×3的超胞,超胞中含有27个Pb和Ti原子,以及81个O原子.由于Cd的有限固溶度,28为了模拟更真实的实验结果,并尽量保持化合物的空间群不发生变化,我们用Sr/Ba/Cd原子替换27个Pb原子中的一个,如图1所示,黄色、绿色、紫色、红色、蓝色、白色的球分别代表了Pb、Sr、Ba、Cd、Ti、O原子.由于计算中所有的原子排布都是周期性的,所以掺杂在超胞(1/3,1/3,1/3)处的原子就相当于替代了27个Pb中的任意一个.接下来,依照实验的晶格常数我们优化了三种化合物的单胞参数.优化分为两步进行,首先固定实验上的单胞参数,优化各原子的坐标;第二步是在第一步的基础上,放松单胞参数和各原子坐标进行优化,优化之后的超胞计算模型及局部放大图如图1所示.分析掺入不同原子的钛酸铅化合物结构可知,优化后的结构仍然保持四方相,自发极化方向仍为c轴,不存在其他铁电相变,这与实验结果保持一致.

图1 3×3×3超胞计算模型及局部放大图Fig.1 3×3×3 supercells calculation models and corresponding enlarged view
3 结果与讨论
接下来,利用优化后的晶格坐标,我们计算了原子自发极化位移δPb、δBa、δSr、δCd、δTi(阳离子对于氧十二面体负电中心的距离),如表1所示.可以看到不同原子的自发极化位移呈现出极大的区别.Sr、Ba的自发极化位移较Pb小,且随着Sr、Ba掺入,化合物的自发极化强度也相应降低,而Cd的自发极化位移高于其他的阳离子,这些结果与实验上的结论16保持一致.从前人的理论分析23得知,自发极化强度反映了铁电性的高低,而阳离子与氧的轨道杂化是铁电性的本质.所以我们需要进一步研究三种化合物不同原子的电子轨道分布.
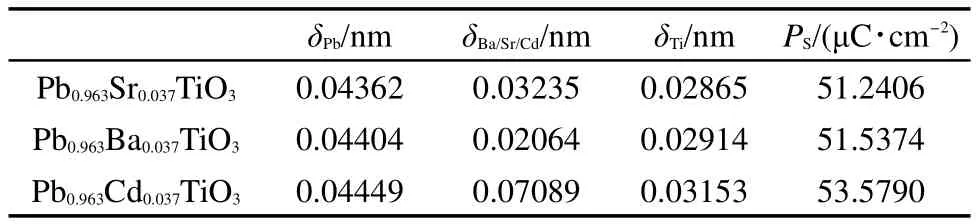
表1 计算的阳离子极化位移(δ)及总体自发极化强度(P S)Table 1 Calculated displacement of cations(δ)and spontaneous polarization(P S)
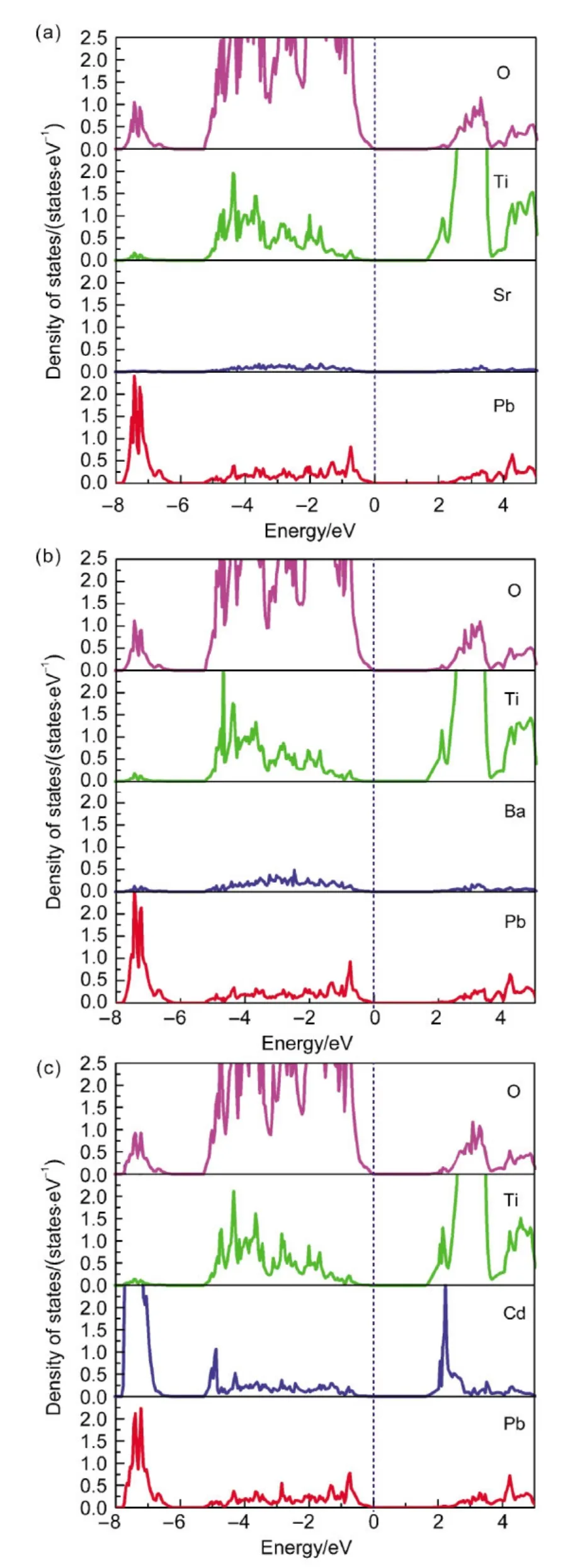
图2 Pb0.963Sr0.037TiO3(a),Pb0.963Ba0.037TiO3(b),Pb0.963Cd0.037TiO3(c)的态密度图Fig.2 Density of states for Pb0.963Sr0.037TiO3(a),Pb0.963Ba0.037TiO3(b),Pb0.963Cd0.037TiO3(c)

图3 三种化合物的(300)面的电子密度分布图Fig.3 Valence electron densities on the(300)plane of three compounds
为了进一步分析内部的电子结构及杂化情况,我们计算了三种化合物的电子态密度.如图2所示.将费米能级固定在0 eV处,可以看到带隙约为1.6 eV.不同原子之间存在很多轨道的重叠,这种重叠说明了不同原子轨道之间的杂化作用.三种化合物在态密度图中有个共同的特征,即Ti和O的态密度峰都很宽,两个轨道的离域性都很强.并且,Ti轨道在O轨道的态密度主峰处有较强贡献,同时O对Ti轨道的态密度主峰处也具有较强的贡献,这种典型的态密度峰“共振”特质表明了Ti轨道和O轨道形成较强的共价键.另外,在价带-7.5 eV附近及-5 eV至价带顶区间,可以看到Cd轨道和O轨道产生的杂化,而在相同的区域,Pb与O也产生了轨道重叠,这说明Pb/Cd原子与O原子之间存在着共价性,这些阳离子轨道与O轨道间的杂化减少了钛酸铅的短程排斥力,进而稳定了铁电结构.29对比这三种化合物可以看出,Cd掺杂钛酸铅的态密度中,Cd轨道与O轨道有效成键,而Ba原子和Sr原子的态密度峰几乎没有与O的态密度峰发生有效重叠,所以认为Ba原子和Sr原子主要以离子形式存在于晶格中.本文中态密度的计算结果与之前的关于无掺杂化合物SrTiO3、BaTiO3、PbTiO3电子结构计算的结果非常吻合,30Sr原子和Ba原子在价带顶部几乎不存在电子轨道的贡献.
因为电子密度分布可以表征原子间的共价程度,所以接下来计算了(300)面电子密度分布,在这个晶面上,可以更容易地比较A位原子与O原子的成键情况.如图3所示,Sr/Ba原子与O的作用更偏于离子性,而Cd―O的共价作用占了主导,且从能量梯度取值可以看到Cd―O之间的共价性要强于Pb―O.为了更好地比较不同位置的共价程度,我们计算了阳离子与氧原子间的最小电子密度.Sr―O最小电荷密度为194.9 e·nm-3,Ba―O最小电荷密度为210.0 e·nm-3,Cd―O最小电荷密度为314.3 e·nm-3.而Pb―O最小电荷密度受不同原子掺杂影响,发生较小的升高或降低,平均最小电荷密度约为286.5 e·nm-3.可知Cd原子与O的共价性要高于其他阳离子,Cd的加入提高了钛酸铅的共价程度.
Cd掺杂PbTiO3是目前所发现的唯一能使PbTiO3负热膨胀性增强的A位掺杂体系,而Cd原子的加入提高了A位原子与O原子之间的共价作用,其他2价原子的加入则降低了A位原子与O原子之间的共价作用,我们试图将共价性和热膨胀联系起来考虑它们之间的关系.实验上分别测试了PbTiO3热膨胀系数为-1.99×10-5°C-1,Pb0.5Ba0.5TiO3热膨胀系数为1.15×10-5°C-1,31Pb0.5Sr0.5TiO3热膨胀系数为-1.11×10-5°C-1,32而 Pb0.94Cd0.06TiO3热膨胀系数为-2.40×10-5°C-1.16Ba原子和Sr原子的掺入,不同程度地削弱了钛酸铅的负热膨胀程度,而Cd的掺入使得化合物负热膨胀程度增强.不难发现Cd原子的掺入促进了A位原子与O的共价作用,提高了低温下的单胞体积,进而提高了负热膨胀程度.而Ba原子和Sr原子的掺入降低了A位原子与O的共价性,进而降低了负热膨胀程度.可以简而言之地说,引入共价性更高的A位原子,会提高A位原子与O的杂化能力,进而提高负热膨胀性,而引入离子性更强的A位原子,会降低A位原子与O的杂化,进而降低负热膨胀程度.再进一步推敲,当A位原子与O的共价作用削弱到一定程度,化合物将不会发生负膨胀,而如大部分物质一样,在温度升高的过程中,体积发生正膨胀.总之,通过第一性原理计算,我们得到了这样的结论:钛酸铅中A位原子与O原子电子轨道杂化促使PbTiO3在室温下保持很高的晶格畸变,它们的共价性不仅是钛酸铅铁电性的起源,也是影响负热膨胀的重要因素.
4 结论
通过结构优化、态密度计算和电子密度计算,可以得到Pb1-xCdxTiO3中Cd原子与O原子轨道存在强烈的杂化,且Cd―O的共价性要强于Pb―O间的共价性,随着Cd含量增加,整体共价作用提高,化合物负热膨胀性增强.而在Pb1-xSrxTiO3和Pb1-xBaxTiO3中,Sr/Ba原子与O的作用更偏于离子性,它们的掺入不同程度地削弱了化合物的共价性,最终影响了负热膨胀性的降低.与实验测量的热膨胀系数对比可以发现,A位原子与O之间的共价性越强,化合物负热膨胀程度越强.而A位原子与氧之间的共价性削弱,负热膨胀程度降低.所以,A位原子与O原子之间的共价性是影响钛酸铅基化合物负热膨胀性的因素.本文理论研究的结果对研究钛酸铅基化合物的负热膨胀性有着深刻的指导意义,为合成具有应用价值的零膨胀化合物提供了理论依据.
(1)Xing,X.R.;Deng,J.X.;Chen,J.;Liu,G.R.Rare Met.2003,22,294.
(2)Chen,J.;Nittala,K.;Forrester,J.S.;Jones,J.L.;Deng,J.;Yu,R.;Xing,X.J.Am.Chem.Soc.2011,133(29),11114.doi:10.1021/ja2046292
(3)Chandra,A.;Pandeya,D.;Mathews,M.D.;Tyagi,A.K.J.Mater.Res.2005,20,350.doi:10.1557/JMR.2005.0062
(4)Evans,J.S.O.Dalton Trans.1999,19,3317.
(6)Zheng,X.G.;Kubozono,H.;Yamada,H.;Kato,K.;Ishiwata,Y.;Xu,C.N.Nat.Nanotechnol.2008,3,724.doi:10.1038/nnano.2008.309
(7)Korcök,J.L.;Katz,M.J.;Leznoff,D.B.J.A m.Chem.Soc.2009,131,4866.doi:10.1021/ja809631r
(8)Zwanziger,J.W.Phys.R ev.B 2007,76,052102.
(9)Greve,B.K.;Martin,K.L.;Lee,P.L.;Chupas,P.J.;Chapman,K.W.;Wilkinson,A.P.J.Am.Chem.Soc.2010,132,15496.doi:10.1021/ja106711v
(10)Goodwin,A.L.;Calleja,M.;Conterio,M.J.;Dove,M.T.;Evans,J.S.O.;Keen,D.A.;Peters,L.;Tucker,M.G.Science 2008,319,794.doi:10.1126/science.1151442
(11)Biernacki,S.;SchefBer,M.Phys.R ev.L ett.1989,63,290.doi:10.1103/PhysRevLett.63.290
(12)Azuma,M.;Chen,W.T.;Seki,H.;Czapski,M.;Olga,S.;Oka,K.;Mizumaki,M.;Watanuki,T.;Ishimatsu,N.;Kawamura,N.;Ishiwata,S.;Tucker,M.G.;Shimakawa,Y.;Attfield,J.P.Nat.Commun.2011,2,347.doi:10.1038/ncomms1361
(13)Pryde,A.K.A.;Hammonds,K.D.;Dove,M.T.;Heine,V.;Gale,J.D.;Warren,M.C.Phase Trans.1997,61,141.doi:10.1080/01411599708223734
(14)Chen,J.;Hu,P.H.;Sun,X.Y.;Sun,C.;Xing,X.R.Ap pl.Phys.L ett.2007,91,171907.doi:10.1063/1.2794742
(15)Chen,J.;Xing,X.R.;Sun,C.;Hu,P.H.;Yu,R.B.;Wang,X.W.;Li,L.H.J.Am.Ch em.S oc.2008,130,1144.doi:10.1021/ja7100278
(16)Chen,J.;Xing,X.R.;Yu,R.B.;Liu,G.R.Appl.Phys.Lett.2005,87,231915.doi:10.1063/1.2140486
(17)Wang,F.F.;Xie,Y.;Chen,J.;Fu,H.G.;Xing,X.R.P hys.Chem.Chem.Phys.2014,16,5237.doi:10.1039/c3cp53197j
(18)Cheng,H.P.;Chen,G.H.;Qin,R.;Dan,J.K.;Huang,Z.M.;Peng,H.;Chen,T.N.,Lei,J.B.Acta Phys.-Chim.Sin.2014,30,281.[程和平,陈光华,覃 睿,但加坤,黄智蒙,彭 辉,陈图南,雷江波.物理化学学报,2014,30,281.]doi:10.3866/PKU.WHXB201312171
(19)Cohen,R.E.;Krakauer,H.F erroelectrics 1992,136,65.doi:10.1080/00150199208016067
(20)García,A.;Vanderbilt,D.Ph ys.Rev.B 1996,54,3817.doi:10.1103/PhysRevB.54.3817
(21)Cohen,R.E.;Sághi-Szabó,G.Phys.Rev.Lett.1998,80,4321.doi:10.1103/PhysRevLett.80.4321
(22)Cockayne,E.;Burton,B.Phys.Rev.B 2004,69,144116.doi:10.1103/PhysRevB.69.144116
(23)Cohen,R.E.Nature 1992,358,136.doi:10.1038/358136a0
(24)Grinberg,I.;Rappe,A.M.Phase Trans.2007,80,351.doi:10.1080/01411590701228505
(25)Yashima,M.;Omoto,K.;Chen,J.;Kato,H.;Xing,X.R.Ch em.Mater.2011,23,3135.doi:10.1021/cm201184y
(26)Kresse,G.;Joubert,D.P hys.Rev.B 1999,59,1758.
(27)Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.L ett.1996,77,3865.doi:10.1103/PhysRevLett.77.3865
(28)Suárez-Sandoval,D.Y.;Davies,P.K.A ppl.Ph ys.Lett.2003,82,3215.doi:10.1063/1.1573362
(29)Kuroiwa,Y.;Aoyagi,S.;Sawada,A.;Harada,J.;Nishibori,E.;Takata,M.;Sakata,M.Ph ys.Rev.Lett.2001,87,217601.doi:10.1103/PhysRevLett.87.217601
(30)Piskunov,S.;Heifets,E.;Eglitis,R.I.;Borstel,G.Comput.Mater.Sci.2004,29,165.doi:10.1016/j.commatsci.2003.08.036
(31)Xing,X.R.;Deng,J.X.;Zhu,Z.Q.;Liu,G.R.J.Alloy.Compd.2003,353,1.doi:10.1016/S0925-8388(02)01178-7
(32)Xing,X.R.;Chen,J.;Deng,J.X.;Liu,G.R.J.A lloy.Compd.2003,360,286.doi:10.1016/S0925-8388(03)00345-1
猜你喜欢
潍坊学院学报(2021年2期)2021-07-22 07:59:12
高等学校化学学报(2021年7期)2021-07-11 16:25:48
原子与分子物理学报(2021年1期)2021-03-29 07:29:40
原子与分子物理学报(2021年1期)2021-03-29 07:28:18
陶瓷学报(2020年2期)2020-10-27 02:16:14
成都信息工程大学学报(2019年6期)2019-08-13 03:31:14
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:34:54
材料科学与工程学报(2016年1期)2017-01-15 13:34:08
贵州师范学院学报(2016年3期)2016-12-01 03:53:53
电源技术(2015年9期)2015-06-05 09:36:03
