
血糖水平对缺血再灌注脑组织过氧化损伤的影响
2014-10-17 09:32成学恭李光来
中国医药导报 2014年24期
任 歆 黄 蕊 成学恭 李光来
煤炭总医院神经内科,北京 100028
脑血管疾病是神经系统的常见病,包括脑出血及脑梗死,是致残率及致死率非常高的疾病,每年可造成数十亿的人群死亡或丧失劳动能力,给社会带来严重的经济负担和社会负担。根据病变血管的不同,可以导致局灶性或弥漫性脑功能缺失,在临床上可以导致患者肢体瘫痪、感觉缺失、言语功能障碍等,给患者的日常生活造成巨大困扰,缺血性脑卒中约占脑卒中的80%,包括脑栓塞、脑血栓形成及腔隙性脑梗死等。早期进行溶栓治疗可以尽可能地恢复阻塞血管的血流,恢复梗死区的血供,以期恢复或改善梗死区的脑功能,但伴随出现却是明显的再灌注损伤问题,后者不仅没有恢复脑卒中造成的神经功能缺失,反而导致病灶局部出现一系列继发性改变,导致脑水肿及神经功能损伤加重[1-2]。大量研究证实,再灌注损伤的机制包括:阻塞血管无复通、微循环障碍、能量匮乏、自由基大量生成、钙超载及诱发一系列有害基因表达等。研究表明,脑缺血急性期可以导致机体应激性血糖升高,血糖水平的异常可以加重脑组织损伤。本实验通过观察不同血糖水平下,缺血再灌注损伤后脑细胞结构、ATP酶的活力及细胞氧化还原能力的变化,对动物模型溶栓过程中血糖水平进行初步探讨。
1 材料与方法
1.1 动物模型制作及分组
取24只健康雄性Wistar大鼠 (2~3月龄,230~260 g),采用急性脑缺血再灌注模型[4-5],按照360 mg/kg腹腔注射10%水合氯醛进行麻醉,动物取仰卧位,双侧逐层分离出颈总动脉,以小动脉夹夹闭造成脑缺血,30 min以后松开动脉夹再灌注2 h,取静脉血并断头处死,1 min内分离出额、颞、顶部大脑皮质,左侧用做病理及电镜检查,右侧置-70℃冰箱保存待测。
1.2 仪器与设备
普通胰岛素与中性鱼精蛋白锌胰岛素(中国徐州万邦生化制药有限公司,江苏)、四乙氧基丙烷(TEP)、硫代巴比妥酸(TBA)等为国产分析纯,黄嘌呤氧化酶、黄嘌呤(美国Sigma公司),超氧化物歧化酶(SOD)标准品(德国BOEHRINGER MANNHEIM公司),ATP测试盒与SOD测试盒(南京建成生物制品研究所),722光栅分光光度计(上海第三分析仪器厂),透射电镜(JEOL-100 型),光镜(OLYMPUS-VANOX 型)。
1.3 分组及给药
实验分组:随机分成4组,每组6只,分别为假手术组(A组),分离出双侧颈总动脉并备线即结束,作为对照;生理盐水对照组(B组);胰岛素(2.1 U/kg)组(C 组);胰岛素(2.1 U/kg)+50%葡萄糖(2 g/kg)组(D组)。参考相关文献资料[3,6],在夹闭双侧颈总动脉造成脑缺血后,B组给予生理盐水4 mL/kg经腹腔内注射;C组给予胰岛素2.1 U/kg(普通胰岛素RI与中性鱼精蛋白锌胰岛素NPH以1∶2的比例分别进行腹腔内注射和皮下注射);D组给予给予胰岛素2.1 U/kg(同C组),同时给予50%葡萄糖注射液2 g/kg腹腔注射。各组于术前0.5 h、术后0.5、1.5、2.5 h取尾血用微量血糖仪(ADVANTAGE,U.S.A)检测血糖。对试验动物以100 W照明灯照射,以保持动物肛温在36.5~37.5℃之间。
1.4 实验方法
1.4.1 脑组织丙二醛(MDA)测定 采用改良八木国夫法[7]。将脑组织制成1%匀浆,取0.3 mL于10 mL小试管中,加入0.7 mL蒸馏水,于漩涡器上混合30 s,加入0.5 mL TBA溶液,用玻璃棒搅拌使液体均匀,放入95℃左右的甘油浴中搅拌,加热60 min后取出,放冷水中冷却,加入2.5 mL正丁醇于漩涡器上提取30 s,再以3000 r/min离心10 min。吸取上清液于1 cm比色皿中,在分光光度计上于535 cm波长处,测定吸光度值。以正丁醇为参比。同时以四乙氧基丙烷标准溶液(1 nmol/mL)1 mL于10 mL小试管中,加入0.5 mL TBA溶液做标准,操作同上。
1.4.2 脑组织SOD测定 采用黄嘌呤氧化酶法。根据试剂盒提供的步骤进行,取脑组织上清液在分光光度计波长550 nm处比色,读取OD值。按照每毫克脑组织蛋白在1 mL反应液中SOD抑制率达50%时所对应的SOD量为一个亚硝酸盐单位(NU)。
1.4.3 脑组织ATP酶活性检测 在体内ATP酶可以将ATP分解生成ADP和无机磷,因此无机磷的含量可间接反映ATP酶活力的状态。根据试剂盒提供的步骤进行,在分光光度计波长660 nm处比色,读取OD值。以每小时每毫克组织蛋白中ATP酶分解ATP产生1 μmol无机磷的量为1个ATP酶活力单位,即微克分子磷/(mg prot·h)。
1.5 脑组织形态学特点
试验动物处死后在1 min内快速取出一侧大脑半球,固定于中性福尔马林液中,常规脱水、包埋后,切成0.3 mm的组织切片,以伊红、苏木精染色,置于OLYMPUS-VANOX型光镜下,观察脑组织形态结构。
每组试验动物中随机抽取2只,处死后快速取额、顶叶大脑组织,投入2.5%的戊二醛溶液中固定,30 min后取出,快速切割为1 mm×1 mm×1 mm的组织块,重新投入固定液,按电镜标本常规脱水、包埋后做超薄切片,切片在JEOL-100型透射电镜下观察。
1.6 统计学方法
采用SPSS 13.0统计学软件进行数据分析,计量资料数据用均数±标准差(±s)表示,多组间比较采用单因素方差分析,组间两两比较采用LSD-t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 各组动物血糖水平比较
按照上述试验方法,A组为假手术组,血糖水平在正常范围内,B组血糖水平由于机体应激明显升高,缺血再灌注1.5 h后升高更为明显(P<0.01),给予胰岛素干预后 (C组)血糖水平明显降低 (P<0.01),与C组等量的胰岛素干预并同时给予适当葡萄糖补充(D组),可将血糖水平控制在正常范围的高限。见表1。
2.2 各组动物缺血再灌注后氧化应激反应指标的比较
大鼠急性脑缺血再灌注后脑组织内MDA、SOD含量变化研究结果表明,缺血再灌注损伤发生后,每100毫克脑组织蛋白内的MDA含量明显升高,SOD含量明显下降,在B组及C组更为明显(P<0.01),而且两组间差异无统计学意义(P>0.05);采用与C组等量胰岛素加葡萄糖干预(D组)将缺血再灌注后血糖水平调节至正常值高限,其MDA含量及SOD含量同B组及C组相比较,差异有统计学意义(P<0.01),提示血糖值在正常范围高限可以减轻氧化应激损伤。见表2。
表1 各组大鼠血糖水平比较(mmol/L,±s)
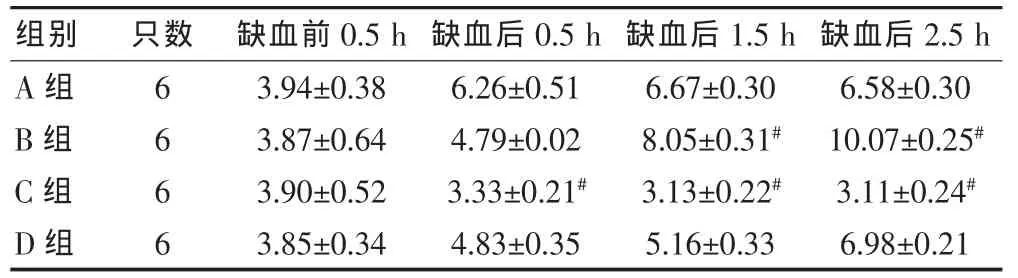
表1 各组大鼠血糖水平比较(mmol/L,±s)
注:与A组相比,#P<0.01
组别 只数 缺血前0.5 h 缺血后0.5 h 缺血后1.5 h 缺血后2.5 h A组B组C组D组6 6 6 6 3.94±0.38 3.87±0.64 3.90±0.52 3.85±0.34 6.26±0.51 4.79±0.02 3.33±0.21#4.83±0.35 6.67±0.30 8.05±0.31#3.13±0.22#5.16±0.33 6.58±0.30 10.07±0.25#3.11±0.24#6.98±0.21
表2 各组大鼠急性脑缺血再灌注后脑组织内MDA、SOD含量(±s)

表2 各组大鼠急性脑缺血再灌注后脑组织内MDA、SOD含量(±s)
注:与 A组比较,#P<0.01;与 D组比较,*P<0.01;MDA:丙二醛;SOD:超氧化物歧化酶
组别 只数 MDA(μmol/100 mg prot) SOD(NU/100 mg prot)A组B组C组D组6 6 6 6 40.42±4.91 59.33±5.59#*52.38±6.69#*44.13±4.27 188.67±20.93 126.12±20.43#*141.53±16.97#*170.80±14.23
2.3 各组动物缺血再灌注后脑组织内ATP酶活性的比较
在急性缺血再灌注损伤发生后,应激性血糖升高(B组)以及胰岛素干预使血糖过度降低(C组)都可以导致脑组织内氧化应激加重,导致ATP酶活性的下降(P<0.01),但两组间差异无统计学意义(P>0.05),而将血糖控制在正常值高限(D组)可以明显升高ATP酶的活性,减轻氧化应激损伤(P<0.01)。见表3。
表3 各组大鼠急性脑缺血再灌注后脑组织内的ATP酶活性比较[微克分子磷/(mg prot·h),±s]
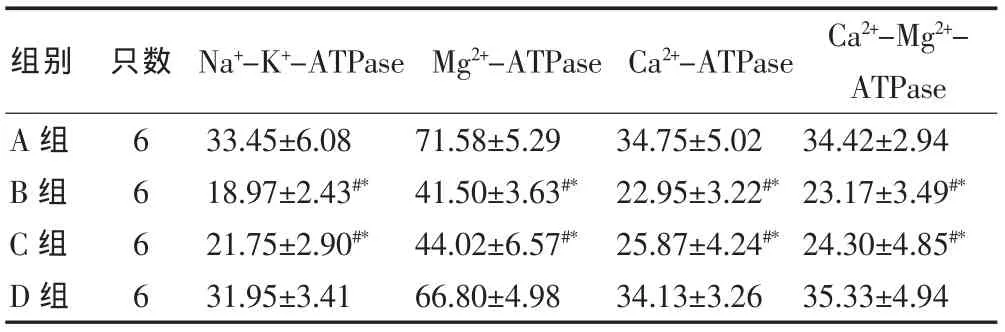
表3 各组大鼠急性脑缺血再灌注后脑组织内的ATP酶活性比较[微克分子磷/(mg prot·h),±s]
注:与A组比较,#P<0.01;与D组比较,*P<0.01
组别 只数 Na+-K+-ATPaseMg2+-ATPase Ca2+-ATPase Ca2+-Mg2+-ATPase A组B组C组D组6 6 6 6 33.45±6.08 18.97±2.43#*21.75±2.90#*31.95±3.41 71.58±5.29 41.50±3.63#*44.02±6.57#*66.80±4.98 34.75±5.02 22.95±3.22#*25.87±4.24#*34.13±3.26 34.42±2.94 23.17±3.49#*24.30±4.85#*35.33±4.94
2.4 脑组织镜下组织学改变
光学显微镜下脑组织切片:A组大脑皮质细胞分层清晰,细胞结构正常;脑缺血30 min,再灌注2 h后,B组与C组皮质细胞层次结构消失,排列紊乱,细胞水肿,胞核皱缩、深染,大部分锥体细胞突起消失,血管周围水肿明显;D组可见大脑皮质细胞仍有分层痕迹,少部分锥体细胞发生轻微肿胀,细胞突起有部分断裂,但细胞内胞核大部分完整。
电镜下超微病理改变:A组皮质细胞结构正常,核膜完整,细胞器及细胞核结构正常;缺血再灌注后,B组与C组皮质细胞呈均质化,核膜呈节段性破坏,核周水肿,线粒体嵴断裂、脱失,基质颗粒脱落、消失,线粒体肿胀,呈烧瓶样或空泡化,数量明显减少,内质网肿胀,核糖体脱落,细胞膜呈节段断裂,细胞突起消失;D组缺血再灌注损伤后病理改变明显减轻,可见脑细胞水肿及血管周围间隙内水肿较轻,皮层细胞层次基本存在,脑细胞内仅有部分线粒体发生肿胀,细胞器膜及核膜损伤较轻。
3 讨论
已有组织形态学研究表明[8],脑缺血再灌注后可以发生严重的组织损伤,主要表现为细胞及细胞器的膜损伤,细胞膜及线粒体膜崩解,核膜出现节段性损伤,内质网空泡化,核糖体脱落,细胞胞浆水肿疏松,细胞突起出现严重水肿甚至消失等。已有证据表明,脑缺血后再灌注可以导致氧自由基呈爆发性增加,并攻击细胞膜及细胞器膜上的不饱和脂肪酸,从而破坏膜的完整性,导致膜功能损伤甚至丧失[9-10]。此外,缺血再灌注损伤还可以导致神经元内DNA、RNA、蛋白质和氨基酸发生交联氧化,多糖分子聚合和氧化,不能合成消除氧自由基以及修复细胞膜所需的蛋白酶,导致脑细胞功能的进一步恶化,从而使再灌注损伤进行性加重。本实验研究表明,脑缺血30 min,再灌注2 h后,脑组织内MDA含量升高,SOD含量降低,ATP酶活力明显降低,脑组织超微结构出现明显病理改变,提示脑缺血再灌注后脑细胞出现严重的氧化应激损伤。
众所周知,线粒体是自由基产生的重要场所。电子漏学说[11-12]解释了氧自由基的生成途径。线粒体通过呼吸链电子漏机制进入超氧自由基代谢途径,生成有害的活性氧(ROS),包括O2-、H2O2等。 呼吸链底物端(泛醌区)及氧端(细胞色素区)均有电子漏出,底物端的电子漏导致线粒体生成O2-、H2O2,并还原成H2O,起到部分清除氧自由基的作用。多条代谢途径的复杂平衡决定了线粒体生成氧自由基的水平。正常生理状态下线粒体产生氧自由基的水平较低,而在脑缺血状态下,由于细胞缺氧,线粒体内呼吸链处于高底物还原状态,此时的细胞色素C大部分游离到胞浆中,有利于大量电子漏出及积聚。缺血后再灌注瞬间给处于高还原状态的呼吸链迅速充氧,使细胞内氧自由基大量急剧生成[13]。在可承受的某一浓度范围内,ROS可以诱导细胞进行胁迫应答,并通过改变许多基因表达来维持能量代谢以保护脑细胞;但当超过其可承受的临界值后,ROS便可诱导线粒体膜渗透性发生改变,导致线粒体肿胀,线粒体功能下降,合成ATP减少,细胞膜上的各种ATP酶活性下降,导致细胞膜内外离子转运失平衡,细胞外膜膜电位降低,作为细胞生死主开关的线粒体通透性转变孔长时间开放,使得线粒体基质膨胀,线粒体外膜破裂,膜间隙中的细胞色素C、细胞凋亡诱导因子(AIF)、Ca2+以及膜间隙中其他凋亡因子被释放至细胞质中。细胞色素C可以通过Apaf-1以级联的方式激活各型胱冬肽酶,而细胞色素C的释放与胱冬肽酶的激活之间又形成一个互相促进的反馈性自我放大回路,启动凋亡程序[14]。线粒体间隙内的AIF是一种57 kD的双功能黄素蛋白,除具有电子供体/受体功能以外,还可以独立作用于核染色质,直接进入细胞核内,独立地将DNA断解为60 kb左右的片段,引起染色体凝缩和DNA的大规模断片化,再灌注损伤可以导致AIF大量释放[15]。同时,Ca2+的释放也可以诱导死亡受体的配体表达,激活Ca2+依赖性酶、核酸酶、磷酸化酶,协助激活胱冬肽酶,激活的这些蛋白再以级联反应作用于细胞内骨架蛋白、转录因子以及细胞周期调节蛋白,或直接作用于核DNA分子,引起细胞凋亡[16]。因此,减少氧自由基的生成,降低其对线粒体的损害,是减轻脑缺血再灌注损伤的重要环节。
近年来,人们在对脑缺血急性期血糖水平的大量研究中发现,脑卒中急性期高血糖的发生率非常高,其形成原因可能是由于脑卒中引起的机体应激性高血糖:脑卒中导致患者下丘脑-垂体轴受到急性刺激,促肾上腺皮质激素分泌异常增多,增多的皮质醇可以导致蛋白分解加剧,糖异生原料增加,同时增加的皮质醇也会抑制葡萄糖的利用,从而间接促进了血糖升高。本实验研究表明,高血糖可以导致机体氧自由基生成增多,ATP酶活性下降,ATP产生途径破坏,钠泵功能障碍,最终导致脑细胞水肿,同时糖原在体内无氧酵解也可加速产生乳酸,大量乳酸堆积也可以导致脑水肿加重和酸中毒,使脑梗死面积扩大,血脑屏障进一步破坏,增加脑出血转化的风险[17-18]。高血糖还可增加血液中血小板的黏附力,减少前列环素的合成,使血液黏滞性增加,脑血管微循环障碍加重,从而导致卒中复发[19]。同时,人们在充分重视并积极治疗高血糖时,临床观察发现低血糖同样危害极大。因为脑细胞无能量贮备,因此对血糖的依赖极大,而脑卒中患者的脑细胞对低血糖的耐受更差,一旦出现低血糖,脑细胞的功能最先受损,并可能导致神经功能的不可逆损害[20]。
本实验从脑组织缺血再灌注后,大鼠体内不同血糖水平对脑细胞氧化还原状态及能量代谢的角度,同时观察脑组织超微病理结构的特点,进一步证实了:高血糖及低血糖水平,均可能导致氧化还原酶的破坏及ATP酶活力的下降,而正常高限的血糖水平下氧化还原损伤最小。提示高血糖及低血糖均可造成严重的缺血再灌注脑损伤。组织超微结构的表现也证明,将血糖水平控制在正常值高限可以明显减轻缺血再灌注脑损伤。
在长期的动物实验研究中[21],局灶性脑缺血及不完全性脑缺血被认为更符合临床缺血性中风,因而血糖水平对不完全性脑缺血再灌注损伤的影响为指导临床研究及治疗提供了新的实验室依据。
[1]罗祖明,董佑忠,彭国光.脑血管疾病治疗学[M].北京:人民卫生出版社,1999:455-462.
[2]HackeW,KasteM,OlsenTS,etal.Acutetreatmentofischemia stroke [J].Cerebrovascular Diseases,2000,10 (supple 3):22-33.
[3]Voll CL,Aner RN.Insulin attenuates ischemic brain damage independent of its hypoglycemic effect[J].J Cereb Blood Flow Metab,1991,11(6):1006-1014.
[4]Macmanus JP,Buchan AM,Hill IE,et al.Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain[J].Neurosci Lett,1993,164:89.
[5]开丽,王中峰.人参二醇组皂甙对缺血再灌注脑组织超微结构及NOS的影响[J].中国应用生理学杂志,1998,14(3):205-207.
[6]王艺东,黄如训,李玲,等.局灶脑缺血后胰岛素对基因表达的影响[J].中风与神经疾病杂志,2000,17(3):144-146.
[7]齐风菊,周玫,陈瑗,等.血浆丙二醛含量测定方法-改良的八木国夫法[J].第一军医大学学报,1986,6(21):152.
[8]Jekins LW,Povlishoke JT,Lewelt W,et al.The role of postischemia recirculation in the development of ischemia neuronal injury following complete cerebral [J].Neuropathol(Berl),1981,5:205-220.
[9]TraystmanRJ,KirschJR,KoehlerRC.Oxygenradicalmechanisms of brain injury following ischemia and reperfusion[J].J Appl Physiol,1991,71(4):1185-1195.
[10]White BC,Rafols JA,De Graca DJ,et al.Fluorescent histochemical localization of lipid peroxidation during reperfusion[J].Ann Emerg Med,1992,21:634.
[11]赵卫华,徐建兴,陈清棠.缺血再灌注期间大鼠线粒体的变化[J].中风与神经疾病杂志,2000,17(3):133-135.
[12]Perez-Pinzon MA,Xu GP,Born J,et al.Cytochrome C is released from mitochondria into the cytosol after cere-bral anoxia or ischemia [J].J Cereb Blood Flow Metab,1999,19:39-43.
[13]Winker SP,Msnoz-Ruizl.Mechanism of action of mannitol[J].Surg Neurosurg,1995,45:59.
[14]Chen Q,Gong B,Almasan A.Distint stages of cytochrome release from mitochondria:Evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis[J].Cell Death Differ,2000,7(2):227-233.
[15]Patterson SD,Spahr CS,Daugas E.Mass spectrometric identification of proteins released from mitochondria undergoing permeability transition [J].Cell Death Differ,2000,7(2):137-144.
[16]Chakraborti T,Das S,Mondal M,et al.Oxidant,mitochondria and calcium:an overview[J].Cell Signal,1999,11(2):77-85.
[17]赵大志.高血糖与急性脑卒中加重相关性分析[J].现代医药卫生,2008,24(20):3095.
[18]余志平.高血糖与脑卒中的临床和预后关系[J].中国现代医生,2008,5(16):151-152.
[19]梁存福.急性脑梗塞患者早期空腹血糖水平与预后的关系[J].西南军医,2010,12(3):417-418.
[20]Shintani S,Tsuruoka S,Shiigai.Hypoglycemia hemiplegia:a repeat SPECT study[J].J Neurol Neurolsury Psychiatry,1993,56:700-701.
[21]Auer RN.Insulin,blood glucose levels,and ischemic brain damage[J].Neurology,1998(supple 3):39-43.
猜你喜欢
青少年科技博览(中学版)(2023年5期)2023-06-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中成药(2017年6期)2017-06-13
新青年(2017年4期)2017-04-15
中国康复理论与实践(2015年10期)2015-12-24
吉林大学学报(医学版)(2015年4期)2015-12-17
吉林大学学报(医学版)(2015年5期)2015-12-16
中国体外循环杂志(2015年3期)2015-12-08
癌变·畸变·突变(2015年3期)2015-02-27
河北医科大学学报(2011年1期)2011-03-25
