
含氧化合物加氢脱氧的研究进展
2014-10-10 03:19:34桑小义李会峰李明丰李大东
石油化工 2014年4期
桑小义,李会峰,李明丰,李大东
(中国石化 石油化工科学研究院,北京 100083)
含氧化合物加氢脱氧的研究进展
桑小义,李会峰,李明丰,李大东
(中国石化 石油化工科学研究院,北京 100083)
简要介绍了生物油中主要含氧化合物的类型,包括酯类、酚类、呋喃类和酮类等含氧化合物,重点介绍了不同类型含氧化合物在加氢脱氧过程中的反应途径及所涉及的化学反应,分析了不同类型催化剂参与下的反应途径和产物选择性存在的差异,并对含氧化合物加氢脱氧的未来研究方向提出了建议。
生物油;含氧化合物;加氢脱氧;酯类; 酚类;呋喃;酮类
随着原油重质化和环境污染的日益加剧,煤液化油、费托合成油和生物油作为理想的可替代能源备受研究者的关注,尤其是生物油,因其具有污染物排放少、循环周期短的特点而成为研究的焦点[1]。与原油相比,生物油中含氧化合物的含量较高,主要的含氧化合物类型包括酚类、呋喃类、酯类和酮类等[2-4]。由于大量含氧化合物的存在,油品的氧含量有时高达50%(w)以上,从而造成生物油具有燃烧热值低、化学性质不稳定、加热易聚合、酸性较强、对设备的腐蚀性较大等不利特点[5-7],严重阻碍了其作为车用燃料直接使用,因此需要通过加氢脱氧(HDO)对生物油进行提质。
本文对生物油中的主要含氧化合物类型和几种主要含氧化合物在HDO反应过程中发生的化学反应进行了综述,并对未来的研究方向进行了展望。
1 含氧化合物的类型
根据原料和加工工艺的不同,生物油可分为3类:1)源于植物油和动物脂肪的动植物油[8];2)生物质高压液化得到的液化油[9];3)生物质快速热解得到的热解油[10]。3种类型的生物油中含氧化合物的含量和种类有较大差异。
动植物油的氧含量一般低于20%(w),主要的含氧化合物为三脂肪酸甘油酯,脂肪酸链长度一般为C14~22,其中C16和C18约占全部脂肪酸的90%[11]。生物质液化油和热解油的氧含量则较高,有些生物质热解油的氧含量接近50%(w),目前已检测出的含氧化合物已超过300多种[12-14],其中典型的含氧化合物类型如图1所示。由图1可看出,生物质热解油中的含氧化合物类型主要包括酸类、酯类、呋喃类、酚类和醛类等,各类含氧化合物以含芳环类含氧化合物为主。不同原料得到的生物质热解油的组成见表1[15-18]。由表1可看出,采用不同原料得到的生物质热解油的组成基本相同,主要成分为水(含量10%~30%(w));由于原料的不同,酚类、呋喃类和酯类等组分的含量有较大差异,其他组分主要包括糖类以及一些很难被检测到的木质素热解成分等。由此可见,生物油中含氧化合物的含量较高,成分复杂,研究各类含氧化合物在HDO过程中发生的化学反应具有重要的意义。
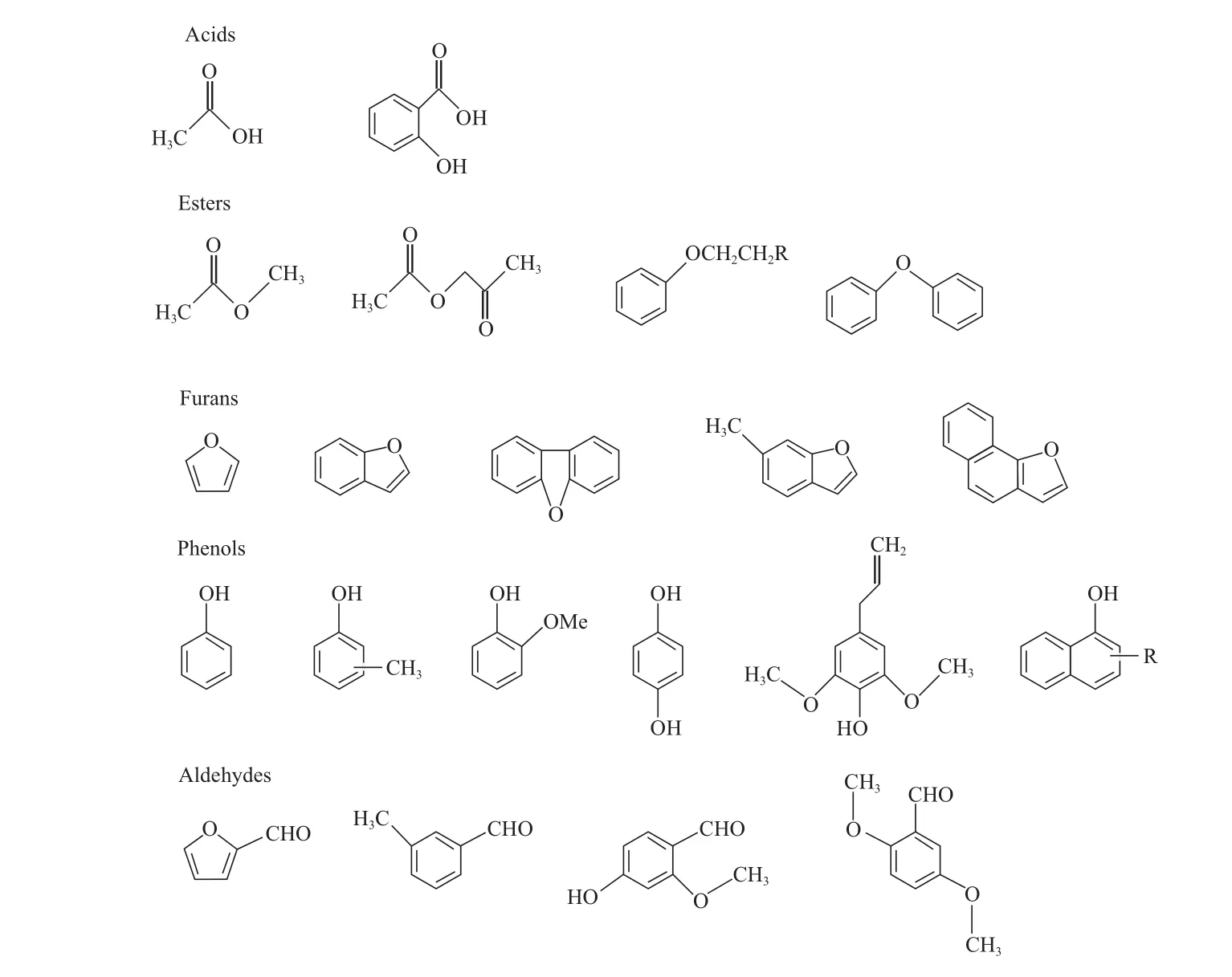
图1 生物质热解油中主要含氧化合物的类型Fig.1 Major oxygenic compounds in biomass pyrolysis oil.
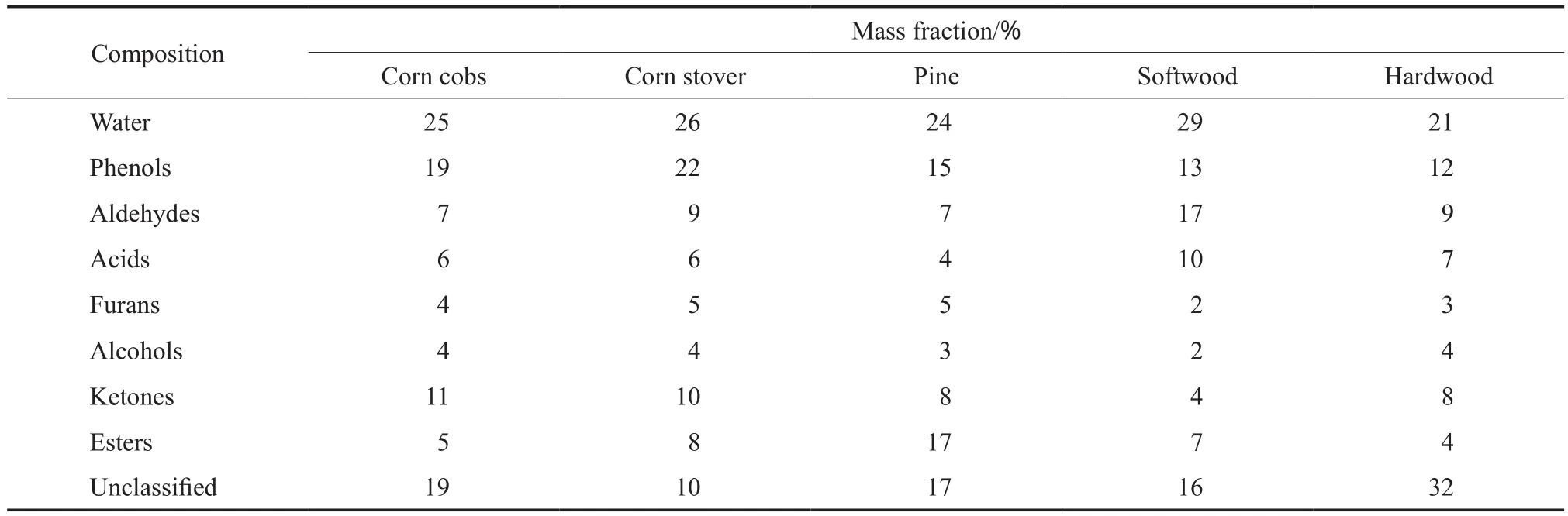
表1 不同原料得到的生物质热解油的组成[15-18]Table 1 Composition of biomass pyrolysis oils from different raw materials[15-18]
2 HDO过程的主要化学反应
2.1 酯类化合物的HDO反应
酯类是动植物油的主要成分,在HDO过程中主要按以下途径进行反应:1)HDO途径,先加氢生成醇,然后再脱水;2)脱羧途径,酯先水解生成醇和酸,酸直接脱羧基以脱除氧,或酸先生成醇类中间产物,然后再脱氧。
Rasmus等[19]在生产绿色柴油的加氢过程中提出,在反应温度350 ℃时,菜籽油在硫化态NiMo/Al2O3催化剂上首先发生的是脂肪酸上双键的加氢饱和,然后按照图2所示的两个不同的反应途径发生脂肪酸和C3骨架的断裂。第1个途径是HDO,生成6 mol H2O、1 mol C3H8和3 mol与脂肪酸链长相同的正构烷烃(n-C18和n-C22);第2个途径是加氢脱羧,生成3 mol CO2、1 mol C3H8和比脂肪酸少一个碳原子的正构烷烃(n-C17和n-C21)。在反应过程中,NiMo/Al2O3催化剂的活性较高,原料脱氧率高达95%以上,产物中加氢脱羧产物的选择性(65%)高于HDO产物的选择性(35%),氢耗也明显较低。
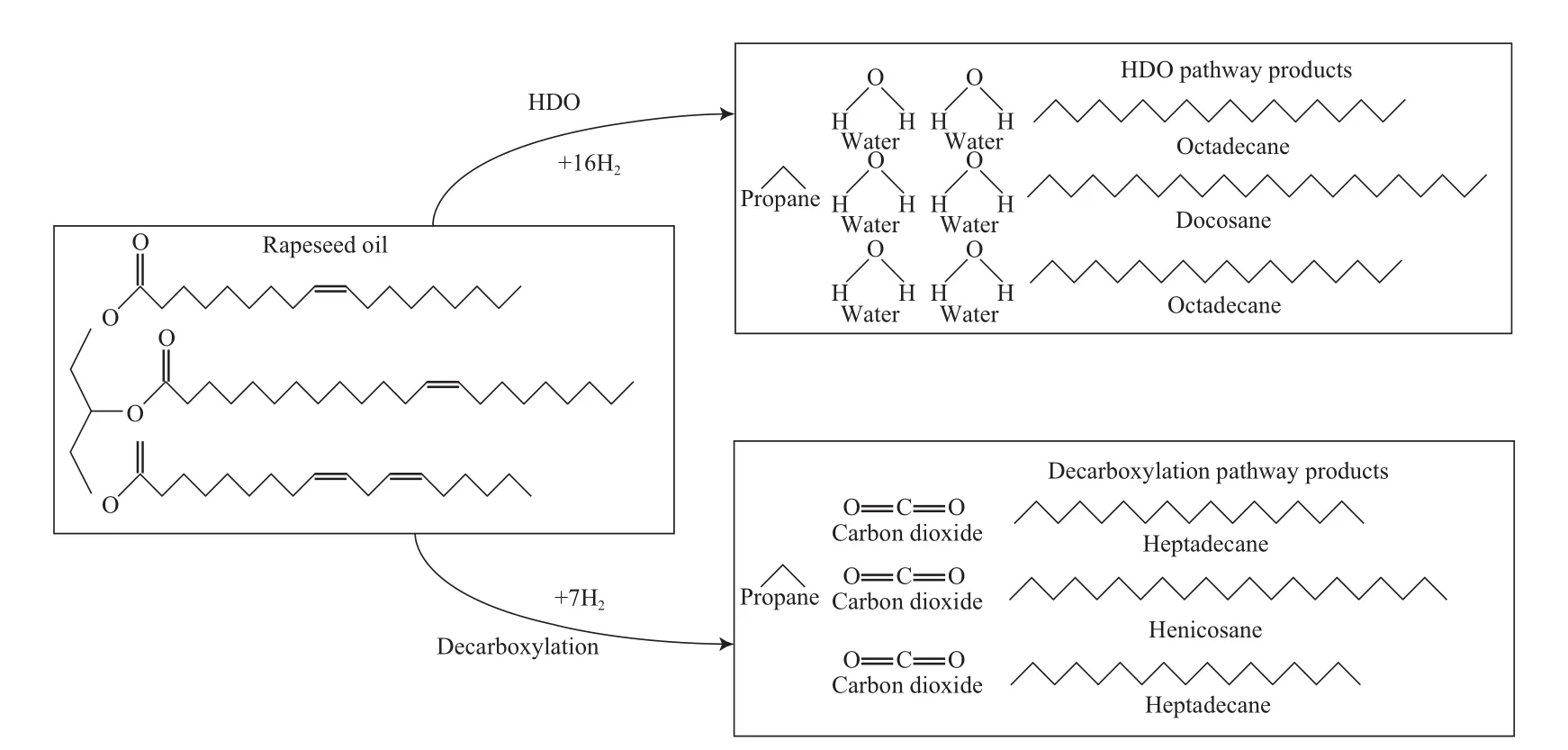
图2 菜籽油的加氢处理反应途径Fig.2 Reaction path for hydrotreating rapeseed oil.
Rasmus等[19]还研究了在300 ℃下,采用硫化态NiMo/Al2O3催化剂催化月桂酸甲酯的加氢反应过程。研究结果表明,反应得到的液体碳氢化合物均为11或12个碳原子,且大多是十二醇、n-C11、n-C12和相应的烯烃,并有少量的十二醛和十二烷酸。这些产物证实了反应有上述两个途径。与加氢脱羧途径相关的产物是C11的烯烃和烷烃,并没有观察到含氧中间物。HDO途径生成的C12机理较复杂,反应的第1步为氢与酯上的氧连接生成醛,然后加氢生成醇、烷烃和水(如图3所示)。反应中烯烃与烷烃的比较高,说明烯烃转化为烷烃的步骤为速率控制步骤。通过进一步研究发现,反应中形成了酮烯醇的平衡(图3方框内),烯醇通过脱水生成烯烃再加氢转化为烷烃。
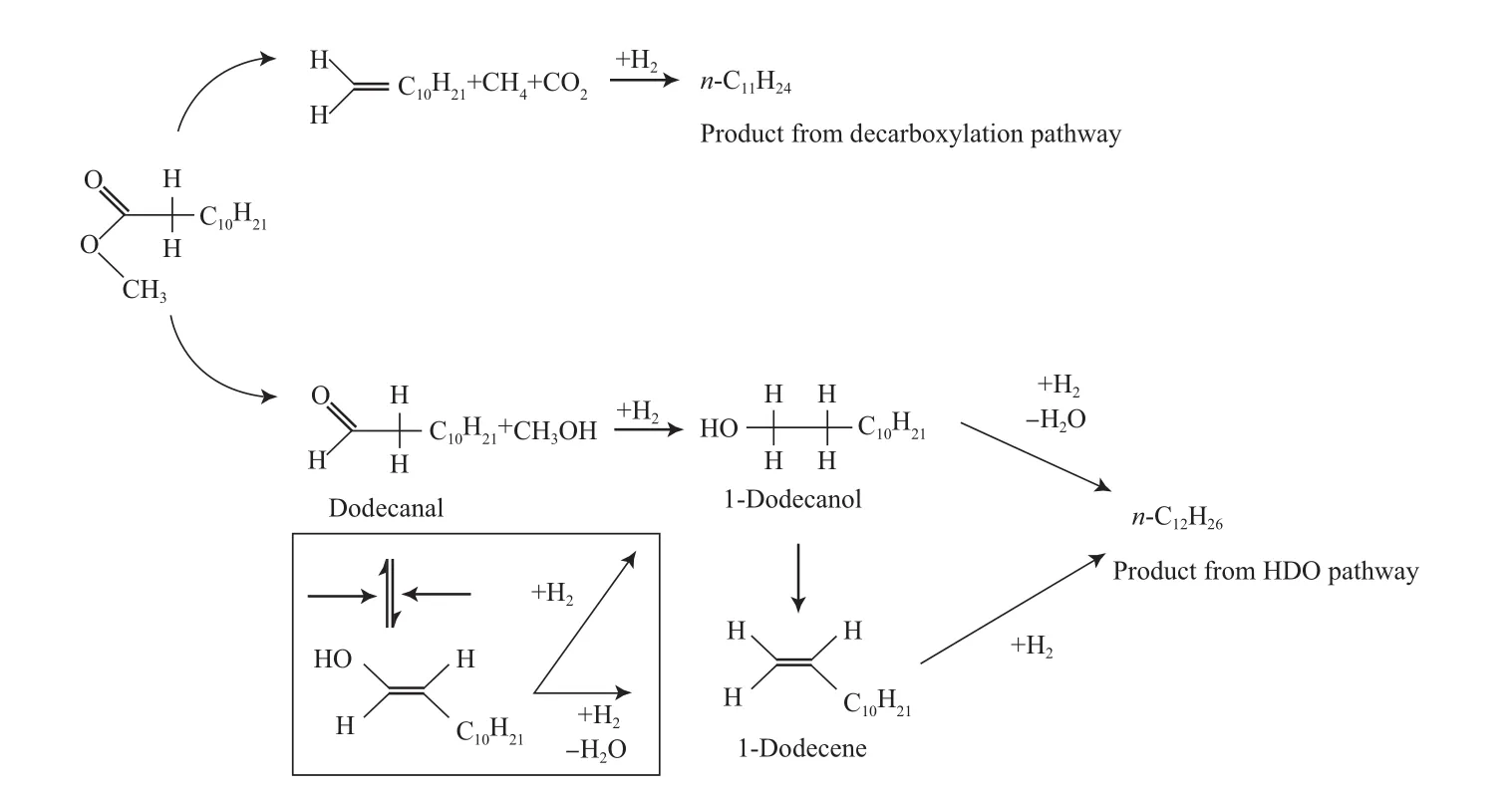
图3 月桂酸甲酯的加氢反应途径Fig.3 Reaction path for the hydrogenation of methyl laurate.
Şenol等[20-21]研究了庚酸甲酯在硫化态NiMo/Al2O3和CoMo/Al2O3催化剂上的加氢反应性能,庚酸甲酯经HDO反应生成C7和C6两种烃类产物,在反应过程中通过加氢和酸催化作用生成醇类、醛类、酸类和酯类的中间产物,反应途径如图4所示。同时,由于催化剂的脱硫作用还生成少量的含硫化合物。在反应过程中,采用NiMo/Al2O3催化剂时庚酸甲酯的脱氧率(约为85%)明显高于采用CoMo/Al2O3催化剂时的脱氧率(约为60%),且NiMo/Al2O3催化剂的加氢活性也高于CoMo/Al2O3催化剂,但氢耗较高。
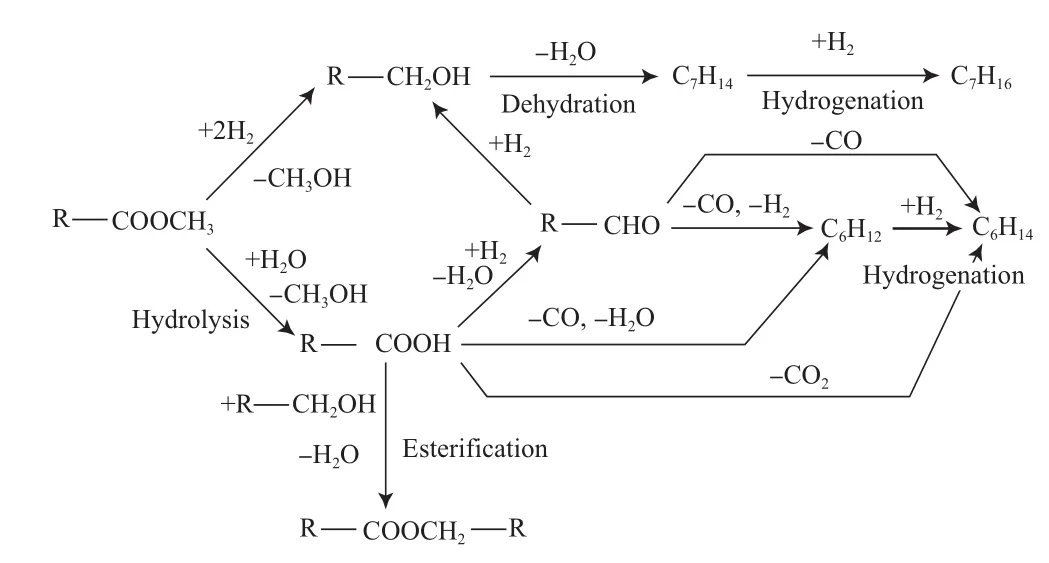
图4 庚酸甲酯在硫化态NiMo/Al2O3和CoMo/Al2O3催化剂上的HDO反应途径(R=C4H9)Fig.4 Reaction path for HDO of methyl heptanoate on sulfurizedNiMo/Al2O3 and CoMo/Al2O3 catalysts(R=C4H9).
酯类化合物采用硫化态催化剂进行HDO反应时,脱氧率较高。在反应过程中不同反应路径得到的产物有所不同,HDO反应途径中氧以H2O的形式脱除,加氢脱羧反应途径中氧以CO2的形式脱除,且加氢脱羧反应得到的脂肪烃比HDO反应得到的脂肪烃少一个碳[22-23];从氢耗角度看,加氢脱羧反应的氢耗较低,更具有低碳环保性。但对反应过程中两种途径所得产物存在差异的根本原因并未指出,有待深入系统的研究。
2.2 酚类化合物的HDO反应
在生物油中,酚类化合物的含量相对较高,尤其是生物质热解油,酚类化合物的含量有时高达50%(w)以上。由于酚类化合物中羟基氧直接与苯环相连,断裂此C—O键所需的活化能比断裂氧与脂肪碳间的化学键所需的活化能高,因此被认为是含氧化合物中最难被脱除的[24]。酚类的HDO反应较为复杂,其中,研究较多的为苯酚和取代苯酚。在有催化剂存在的条件下,酚类的HDO反应主要有两种途径[25]:1)氢化-氢解途径,芳环先加氢生成中间产物环己醇,然后发生消除反应脱除氧;2)直接氢解途径,C—O键直接断裂以脱除氧。
Odebunmi等[26-28]分别采用老化的和新鲜的硫化态CoMo/Al2O3催化剂对甲基苯酚的HDO过程进行了研究。在498~548 K的较低温度范围内,新鲜的硫化态CoMo/Al2O3催化剂上甲基苯酚HDO反应的产物主要是甲苯,甲苯可继续加氢生成甲基环己烷;在623~674 K的较高温度范围内,老化的硫化态CoMo/Al2O3催化剂上,甲基苯酚先吸附在催化剂表面,然后直接反应生成甲基环己烷,不同取代位甲基苯酚的HDO反应顺序为:间甲基苯酚>对甲基苯酚>邻甲基苯酚。
Furimsky等[29]采用CoMo/Al2O3催化剂,对邻位和对位取代苯酚的HDO反应进行了研究。研究结果表明,邻位取代苯酚的HDO反应活性最弱,与其他取代苯酚相比具有较强的脱烷基作用;苯酚、对乙基苯酚和对叔丁基苯酚HDO反应的转化率基本相同。邻位取代苯酚的HDO反应主要按两种途径进行:1)苯酚直接脱氧;2)苯酚先加氢然后再脱氧。反应按途径2进行时,可能会发生消去反应脱除水生成中间产物甲基环己烯,然后甲基环己烯迅速加氢生成甲基环己烷。邻位、对位取代苯酚的HDO反应网络见图5和图6。
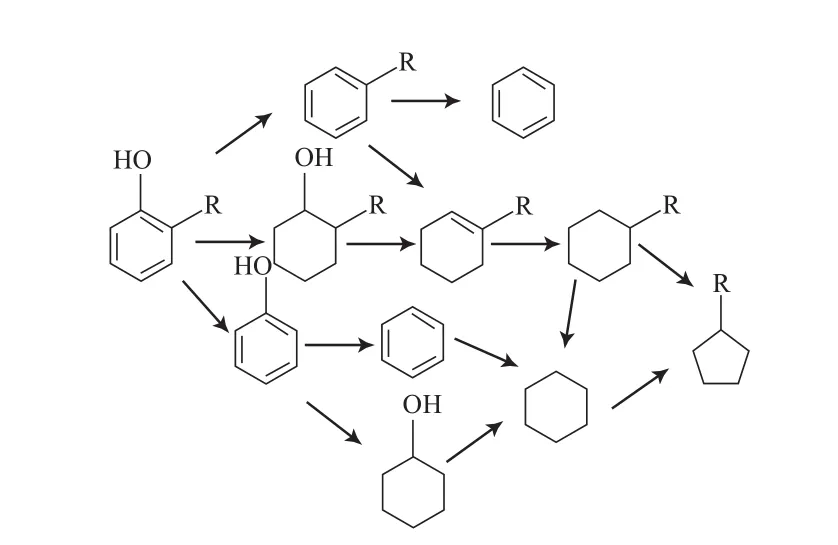
图5 邻位取代苯酚的HDO反应路径Fig.5 Reaction path for HDO of ortho-substituted phenol.
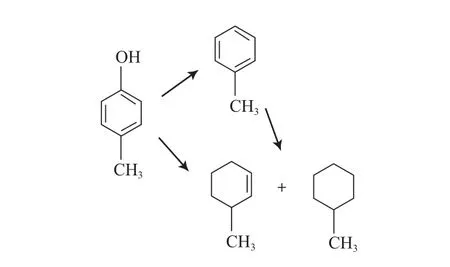
图6 对甲基苯酚的HDO反应路径Fig.6 Reaction path for HDO of 4-methylphenol.
Bui等[30-31]研究了负载在Al2O3,TiO2,ZrO2上的硫化态Mo和CoMo催化剂催化邻甲氧基苯酚的HDO反应,反应途径如图7所示。采用Mo催化剂,载体为Al2O3时,邻甲氧基苯酚HDO反应的主要产物为邻苯二酚;载体为TiO2时,转化率提高近一倍;载体为ZrO2时,原料的脱氧率较高。加入助剂Co后,邻甲氧基苯酚氢解速率加快,先脱甲氧基生成苯酚,再直接脱氧生成苯。采用CoMo/ZrO2催化剂时脱氧率约为80%,产物中含有大量的苯;而采用CoMo/TiO2和CoMo/Al2O3催化剂时脱氧率约为60%,产物中生成的苯和环己烯的量基本相同。在这3种催化剂中CoMo/ZrO2催化剂的氢解活性最高,可使邻甲氧基苯酚直接脱氧生成苯。
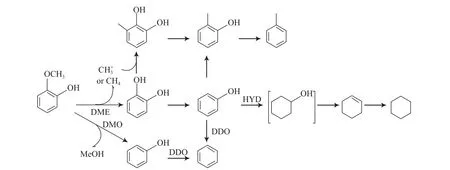
图7 邻甲氧基苯酚在硫化态CoMo催化剂上的HDO反应网络Fig.7 Reaction network for HDO of o-methoxyphenol on sulfurized CoMo catalysts.
Lin等[32]研究了Rh基催化剂上邻甲氧基苯酚的HDO反应过程。研究结果表明,邻甲氧基苯酚在Rh基催化剂上是按先加氢后脱氧的途径进行反应,反应过程见图8。邻甲氧基苯酚先发生加氢反应得到中间产物邻甲氧基环己酮和邻甲氧基环己醇,再脱甲氧基和羟基生成环己烷(选择性达45%)。
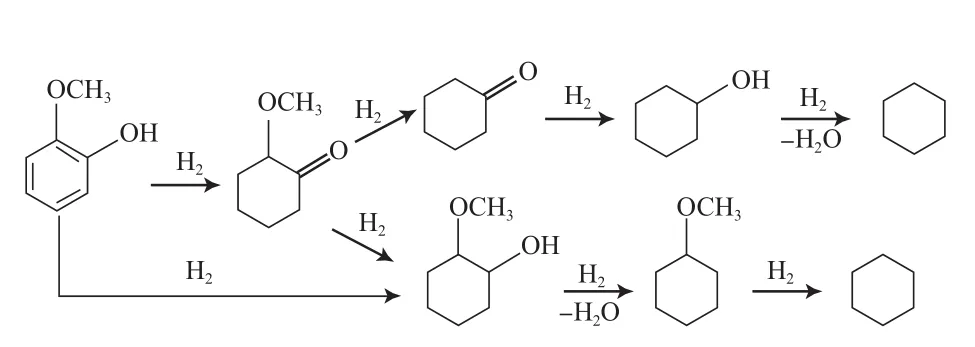
图8 邻甲氧基苯酚在Rh基催化剂上的HDO反应途径Fig.8 Reaction path for HDO of o-methoxyphenol on Rh-based catalysts.
王威燕等[33]研究了270 ℃下非晶态催化剂La-Ni-Mo-B催化4-甲基苯酚的HDO反应(见图9)。在整个反应过程中,没有检测到甲苯的生成,这说明在La-Ni-Mo-B催化下,4-甲基苯酚没有发生直接脱氧反应,而是先加氢生成环己醇,再脱氧生成甲基环己烷,即按照氢化-氢解的途径进行反应。
从上述研究结果可看出,酚类化合物在硫化态催化剂上进行HDO反应时,反应产物以直接脱氧产物为主(氢解途径),氢耗较低;而在贵金属催化剂和非晶态催化剂上进行反应时,产物则以HDO产物为主(氢化-氢解途径),氢耗较高。徐春华[34]研究了不同反应温度和压力下邻甲酚在硫化态CoMo/Al2O3催化剂上的HDO性能,反应中直接脱氧产物甲苯的选择性高达90%,验证了上述观点。因此,可以从目标产物的特点和氢耗上考虑,选取较适宜的催化剂进行HDO反应。
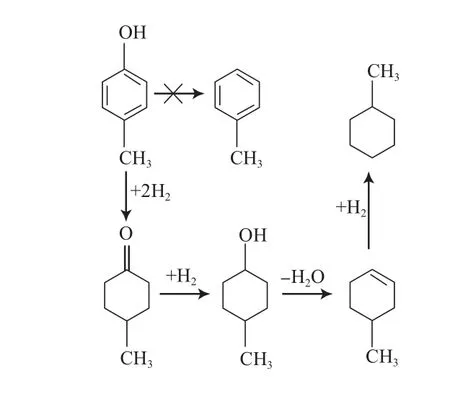
图9 非晶态催化剂La-Ni-Mo-B催化4-甲基苯酚的HDO反应路径Fig.9 Reaction path for HDO of 4-methylphenol on amorphous La-Ni-Mo-B catalysts.
2.3 呋喃类化合物的HDO反应
呋喃类化合物在生物油中的含量也较高,有代表性的模型化合物为四氢呋喃、呋喃和苯并呋喃等[35-37]。Furimsky[36]研究了四氢呋喃的HDO反应过程,提出以下两种反应机理:1)四氢呋喃先发生分子内H原子的迁移,再脱水生成C4H6;2)四氢呋喃先在分子内H作用下发生C—O键断裂形成C—O—催化剂的过渡状态,然后发生C—O键断裂生成C4H8,或C—C键断裂生成C3H6,反应过程如图10所示。
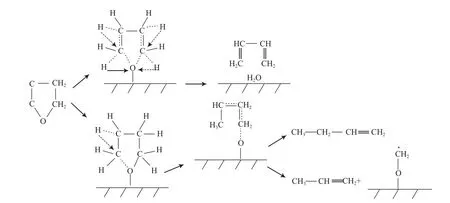
图10 四氢呋喃的HDO反应网络Fig.10 Reaction network for HDO of tetrahydrofuran.
Kreuzer等[37]研究了Pt/Al2O3,Pt/SiO2,Pt/TiO2催化剂对四氢呋喃HDO反应的影响。研究结果表明,3种催化剂HDO反应活性的高低顺序为:Pt/Al2O3>Pt/SiO2>Pt/TiO2,反应过程中四氢呋喃先发生C—O键断裂生成丁醇,然后按两种不同的途径进行反应(见图11):1)丁醇进一步脱氧生成C4H10;2)丁醇发生脱羰反应生成CO和C3H8。反应产物受反应时间和温度的影响较大,当温度为473 K时,产物中C3H8和C4H10的选择性基本相同,但随反应时间的延长,生成较多的C3H8和CO;当温度低于473 K时,产物中含有少量的CH4;当温度高于473 K时,产物中CH4和C3H8的比例基本相同。
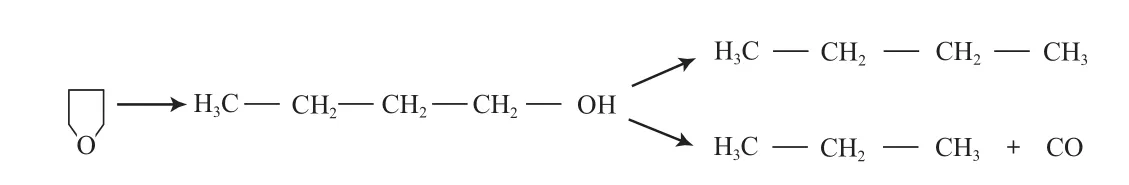
图11 Pt催化剂上四氢呋喃的HDO反应途径Fig.11 Reaction path for HDO of tetrahydrofuran on Pt catalysts.
Bunch等[38-39]研究了硫化态和还原态NiMo/Al2O3催化剂上苯并呋喃的HDO反应网络(见图12)。研究结果表明,在硫化态NiMo/Al2O3催化剂上,只发现一种加氢含氧中间产物2,3-二氢苯并呋喃,反应得到约50%的乙基苯酚及少量的乙苯和乙基环己烷;在原料中加入H2S时,乙基苯酚不能经过氢解生成乙苯,从而导致HDO反应速率下降。在还原态NiMo/Al2O3催化剂上,苯并呋喃经HDO反应生成含氧中间产物的类型较多(如六氢苯并呋喃、八氢苯并呋喃和2-乙基环己醇),中间产物再脱氧生成乙基环己烯和乙基环己烷,产物中并没有发现乙基苯酚和乙苯。
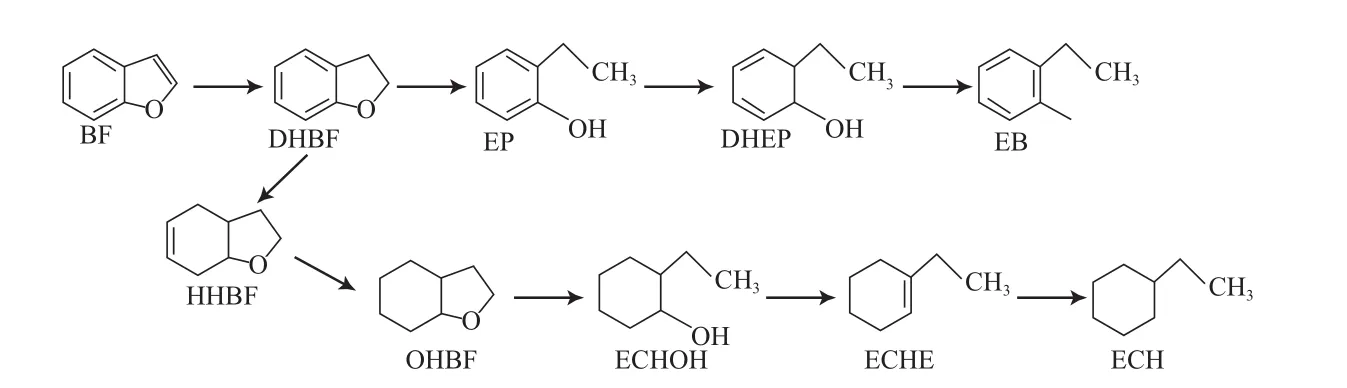
图12 硫化态和还原态NiMo催化剂上苯并呋喃的HDO反应网络Fig.12 Reaction network for HDO of benzofuran on sulfurized and reduced NiMo catalysts.
刘春艳[40]研究了二苯并呋喃在贵金属Pd和Pt催化剂上的HDO反应,主要按照以下3种途径(见图13)进行:1)直接脱氧途径,二苯并呋喃直接脱氧生成联苯;2)氢解途径,先发生C—O键断裂生成苯基苯酚,再加氢生成单环产物;3)加氢途径,首先芳环加氢饱和生成含氧中间产物,然后C—O键断裂生成单环烃。研究结果表明,二苯并呋喃在反应过程中的主要产物为单环烃和联环己烷(选择性约为60%),联苯的选择性较低,且随反应时间的延长,单环烃和联环己烷的选择性逐渐增大,说明二苯并呋喃在反应过程中经历了杂环饱和继而C—O键断裂的过程。
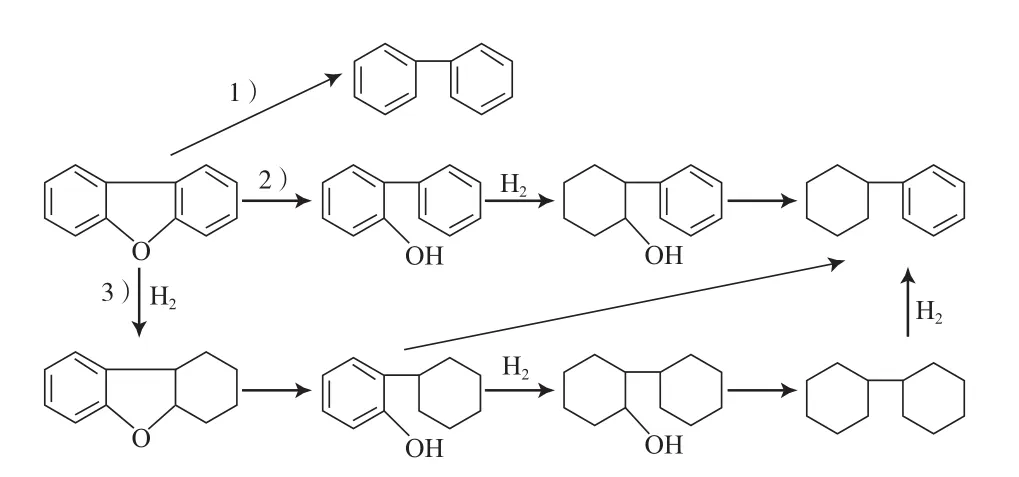
图13 二苯并呋喃的HDO反应路径Fig.13 Reaction path for HDO of diphenylene-oxide.
呋喃类与酚类化合物的HDO反应过程相似,在硫化态催化剂上进行HDO反应时,直接脱氧产物的选择性较高(氢解途径);而在贵金属催化剂和还原态催化剂上反应时,HDO产物的选择性较高(氢化-氢解途径)。由此可见,可根据目标产物的不同,选择不同类型的催化剂进行反应。
2.4 酮类化合物的HDO反应
酮类主要有两种HDO反应途径:1)直接氢解生成烃类化合物;2)先加氢生成醇,再氢解生成烃类化合物(见图14)[25]。
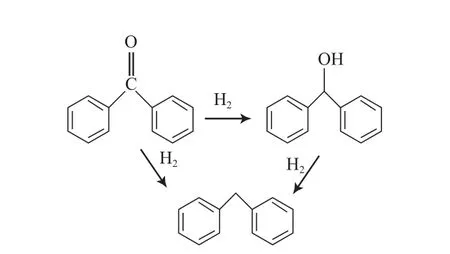
图14 二苯甲酮的HDO反应途径Fig.14 Reaction path for HDO of diphenyl ketone.
Durand等[41]研究了250 ℃下环己酮在硫化态NiMo/Al2O3催化剂上的HDO反应,在反应过程中环己酮先加氢生成环己醇,再脱除氧生成环己烷。杨彦松等[42]研究了非晶态La-Ni-Mo-B和La-Co-Mo-B催化剂催化苯乙酮的HDO反应性能,在反应温度225~275 ℃内,苯乙酮的转化率和脱氧率可达100%,得到的产物为乙基环己烷和乙苯。两种催化剂上产物的选择性不同,采用La-Ni-Mo-B催化剂,当温度高于250 ℃时,产物中乙基环己烷的选择性逐渐降低;而采用La-Co-Mo-B催化剂时,乙基环己烷的选择性则逐渐增大。
3 结语
在含氧化合物的HDO反应过程中,由于采用的催化剂类型不同,反应途径和产物的选择性也有所不同。采用硫化态催化剂时,直接脱氧产物的选择性较高;采用贵金属和还原态催化剂时,HDO产物的选择性较高。目前,研究者大多是针对油品中某种单一含氧化合物进行HDO研究,且未指出采用不同类型的催化剂所得产物选择性有较大差异的本质原因。以后可从以下几个方面进行研究和改进:1)根据不同含氧化合物的化学反应特点,选择较佳的HDO催化剂,以提高HDO转化率和目标产物的选择性;2)进一步详细研究含氧化合物在硫化态、还原态和贵金属等不同类型催化剂上的HDO反应机理,寻找导致反应产物选择性不同的根本原因;3)通过调变载体的比表面积、孔径和酸性来提高催化剂氢解途径产物的选择性,以降低氢耗;4)研究生物油中不同类型含氧化合物在HDO过程中的相互作用,为实际油品的HDO反应和工业化生产提供依据。
[1] 陈满英. 镍基催化剂加氢脱氧性能的研究[D]. 广州:暨南大学,2005.
[2] Zhang Suping,Yan Yongjie,Li Tingchen,et al. Upgrading of Liquid Fuel from the Pyrolysis of Biomass[J].Bioresour Technol,2005,96(5):545 - 550.
[3] Wildschut J,Mahfud F H,Venderbosch R H,et al.Hydrotreatment of Fast Pyrolysis Oil Using Heterogeneous Noble-Metal Catalysts[J].Ind Eng Chem Res,2009,48(23):10324 - 10334.
[4] 魁彦萍,曹晶,周赫,等. 生物油的成分分析及物性测定[J]. 化工生产与技术,2010,17(5):52 - 54,72.
[5] 李其义,万磊,张素平,等. 生物油低温加氢脱氧的研究[J]. 石油化工,2011,40(9):954 - 958.
[6] 武景丽,汪丛伟,阴秀丽,等. 生物油分离方法的研究进展[J]. 石油化工,2008,37(1):95 - 99.
[7] Mortensen P M,Grunwaldt J D,Jensen P A,et al. A Review of Catalytic Upgrading of Bio-Oil to Engine Fuels[J].Appl Catal,A,2011,407(1/2):1 - 19.
[8] 薛颖. 动植物油加氢法生产生物柴油的催化剂和工艺研究[D]. 东营:中国石油大学,2009.
[9] 胡见波,杜泽学,闵恩泽. 生物质高压液化制生物油的影响因素[J]. 石油化工,2012,41(3):347 - 353.
[10] 董芃,齐国利,王丽,等. 生物质快速热解制取生物质油[J]. 太阳能学报,2007,28(2):223 - 226.
[11] 赵阳,吴佳,王宣,等. 植物油加氢制备高十六烷值柴油组分研究进展[J]. 化工进展,2007,26(10):1391 - 1394.
[12] Angelici R J. An Overview of Modeling Studies in HDS,HDN and HDO Catalysis[J].Polyhedron,1997,16(18):3073 - 3088.
[13] 郑小明,楼辉. 生物质热解油品位催化提升的思考和初步进展[J]. 催化学报,2009,30(8):765 - 769.
[14] 朱锡锋,陆强,郑冀鲁,等. 生物质热解与生物油的特性研究[J]. 太阳能学报,2006,27(12):1285 - 1289.
[15] 常胜,赵增立,张伟,等. 不同种类生物油化学组成结构的对比研究[J]. 燃料化学学报,2011,39(10):746 - 753.
[16] 刘运权,龙敏南. 几种不同生物质的快速热解[J]. 化工进展,2010,29(S1):126 - 132.
[17] 王丽红,贾官臣,柏雪源,等. 生物质热解生物油的成分分析[J]. 太阳能学报,2009,30(8):1124 - 1128.
[18] 张琦,常杰,王铁军,等. 生物质裂解油的性质及精制研究进展[J]. 石油化工,2006,35(5):493 - 498.
[19] Rasmus E,Lars S,Per Z,et al. Hydrotreating in the Production of Green Diesel[J].Digital Re fi ning,2010(4):1 - 11.
[20] Şenol O İ,Ryymin E M,Viljava T R,et al. Reactions of Methyl Heptanoate Hydrodeoxygenation on Sulphided Catalysts[J].J Mol Catal A:Chem,2007,268(1/2):1 - 8.
[21] Şenol O İ,Viljava T R,Krause A O I. Hydrodeoxygenation of Aliphatic Esters on Sulphided NiMo/γ-Al2O3and CoMo/γ-Al2O3Catalyst:The Effect of Water[J].Catal Today,2005,106(1/4):186 - 189.
[22] 冯锡兰,柳云骐,陈为超,等. 辛癸酸甘油酯催化加氢脱氧反应规律[J]. 中国石油大学学报:自然科学版,2009,33(5):144 - 147.
[23] 王东军,刘红岩,刘玉香,等. 生物燃料加氢脱氧催化剂的研究进展[J]. 石油化工,2012,41(10):1214 - 1219.
[24] Echeandia S,Arias P L,Barrio V L,et al. Synergy Effect in the HDO of Phenol over Ni-W Catalysts Supported on Active Carbon:Effect of Tungsten Precursors[J].Appl Catal,B,2010,101(1/2):1 - 12.
[25] 王威燕,杨运泉,童刚生,等. 生物油加氢脱氧研究进展[J]. 工业催化,2009,15(5):7 - 14.
[26] Odebunmi E O,Ollis D F. Catalytic Hydrodeoxygenation:Ⅱ. Interactions Between Catalytic Hydrodeoxygenation of M-Cresol and Hydrodesulfurization of Benzothiophene and Dibenzothiophene[J].J Catal,1983,80(1):65 - 75.
[27] Odebunmi E O,Ollis D F. Catalytic Hydrodeoxygenation:Ⅲ. Interactions Between Catalytic Hydrodeoxygenation ofm-Cresol and Hydrodenitrogenation of Indole[J].J Catal,1983,80(1):76 - 89.
[28] Odebunmi E O,Ollis D F. Catalytic Hydrodeoxygenation:Ⅰ.Conversions ofo-,p-,andm-Cresols[J].J Catal,1983,80(1):56 - 64.
[29] Furimsky E. Catalytic Hydrodeoxygenation[J].Appl Catal,A,2000,199(2):147 - 190.
[30] Bui V N,Laurenti D,Delichère P,et al. Hydrodeoxygenation of Guaiacol:PartⅡ:Support Effect for CoMoS Catalysts on HDO Activity and Selectivity[J].Appl Catal,B,2011,101(3/4):246 - 255.
[31] Bui V N,Laurenti D,Afanasiev P,et al. Hydrodeoxygenation of Guaiacol with CoMo Catalysts:PartⅠ:Promoting Effect of Cobalt on HDO Selectivity and Activity[J].Appl Catal,B,2011,101(3/4):239 - 245.
[32] Lin Yuchuan,Li jialiang,Wan Haopeng,et al. Catalytic Hydrodeoxygenation of Guaiacol on Rh-Based and Sulfided CoMo and NiMo Catalysts[J].Energy Fuels,2011,25(3):890 - 896.
[33] 王威燕,杨运泉,罗和安,等. La-Ni-Mo-B非晶态催化剂的制备、加氢脱氧性能及失活研究[J]. 燃料化学学报,2011,39(5):367 - 372.
[34] 徐春华. 加氢脱氧反应对硫化态催化剂结构的影响[D]. 北京:石油化工科学研究院,2011.
[35] Edelman M C,Maholland M K,Baldwin R M,et al. Vapor-Phase Catalytic Hydrodeoxygenation of Benzofuran[J].J Catal,1988,111(2):243 - 253.
[36] Furimsky E. Mechanism of Catalytic Hydrodeoxygenation of Tetrahydrofuran[J].Ind Eng Chem Prod Res Dev,1983,22(1):31 - 34.
[37] Kreuzer K,Kramer R. Support Effects in the Hydrogenolysis of Tetrahydrofuran on Platinum Catalysts[J].J Catal,1997,167(2):391 - 399.
[38] Bunch A Y,Ozkan U S. Investigation of the Reaction Network of Benzofuran Hydrodeoxygenation over Sul fi ded and Reduced Ni-Mo/Al2O3Catalysts[J].J Catal,2002,206(2):177 -187.
[39] Bunch A Y,Wang Xueqin,Ozkan U S. Hydrodeoxygenation of Benzofuran over Sul fi ded and Reduced Ni-Mo/γ-Al2O3Catalysts:Effect of H2S[J].J Mol Catal A:Chem,2007,270(1/2):264 - 272.
[40] 刘春艳. 负载型Pt、Pd及Pt-Pd催化剂上苯并呋喃加氢脱氧反应[D]. 大连:大连理工大学,2012.
[41] Durand R,Geneste P,Moreau C,et al. Heterogeneous Hydrodeoxygenation of Ketones and Alcohols on Sul fi ded NiOMoO3/γ-Al2O3Catalyst[J].J Catal,1984,90(1):147 - 149.
[42] 杨彦松,彭会左,杨运泉,等. La-Ni(Co)-Mo-B非晶态催化剂催化苯甲醛、苯乙酮加氢脱氧反应[J]. 化工进展,2012,31(12):2666 - 2671.
(编辑 王 萍)
Progresses in Researches on Hydrodeoxygenation of Oxygenic Compounds
Sang Xiaoyi,Li Huifeng,Li Mingfeng,Li Dadong
(SINOPEC Research Institute of Petroleum Processing,Beijing 100083,China)
The main types of oxygenic compounds in bio-oil,namely esters,phenols,furans,ketones etc.,were briefly described first. Then the reaction paths and chemical reactions for the hydrodeoxygenation of the oxygenic compounds were emphasized. The differences of the reaction paths and the product selectivity when different types of catalysts were used in the reaction processes were analyzed. Researches on the hydrodeoxygenation reaction paths of the oxygenic compounds were reviewed,and the further research directions were also proposed.
bio-oil;oxygenic compounds;hydrodeoxygenation;esters;phenols;furans;ketones
1000 - 8144(2014)04 - 0466 - 08
TQ 517.4
A
2013 - 11 - 14;[修改稿日期]2014 - 01 - 09。
桑小义(1985—),女,河北省石家庄市人,博士生,电话 010 - 82368083,电邮 sangxy.ripp@sinopec.com。联系人:李明丰,电话 010 - 82368907,电邮 limf.ripp@sinopec.com。
国家高技术研究发展计划项目(2012AA051803)。
猜你喜欢
中学化学(2022年5期)2022-06-17 16:51:48
云南化工(2020年11期)2021-01-14 00:50:54
中国特种设备安全(2019年3期)2019-04-22 05:05:38
中学生数理化·高二版(2016年3期)2016-12-26 09:42:53
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
山东工业技术(2016年15期)2016-12-01 05:30:43
合成化学(2015年4期)2016-01-17 09:01:27
环境科技(2015年5期)2015-11-08 12:08:58
河南科技(2014年15期)2014-02-27 14:12:38
化工生产与技术(2014年6期)2014-02-27 13:42:07
