
四氢异喹啉生物碱jorumycin的全合成研究进展
2014-09-17 06:08陈瑞蛟
济宁医学院学报 2014年1期
陈瑞蛟
(济宁医学院基础学院,山东 济宁 272067)
具有抗肿瘤活性的四氢异喹啉类生物碱(tetrahydroisoquinoline antitumor antibiotics),是天然产物中一类非常重要的化合物。它们具有显著的抗肿瘤、抗菌等活性,在化学、生物学、医学等研究领域受到广泛关注[1]。1974年,加拿大科学家Kluepfel等[2]从土壤来源的葡萄牙链霉菌 Streptomyces lusitanus AYB-1206中分离得到了首例具有抗癌活性的四氢异喹啉类生物碱 naphthyridinomycin。迄今为止,该家族已报道的成员有60余个。其中多个化合物及其衍生物显示了很好的抗癌活性,如乳腺癌、直肠癌、肺癌等。
1 四氢异喹啉生物碱jorumycin的分离及活性
2000年,Fontana等[3-4]从太平洋裸鳃亚目动物Jorunna funebris的皮和黏液中分离得到1个新的双四氢异喹啉生物碱--jorumycin。它与之前分离的renieramycins类生物碱的区别在于C1位上的侧链由当归酰基变为乙酰基。他们研究发现jorumycin具有很好的抗癌活性,结果见表1。

表1 Jorumycin的癌细胞毒性实验
注:P388:老鼠的淋巴腺瘤;A549:人类的肺癌;HT29:人类的结肠癌;MEL28:人类的黑色素瘤
鉴于四氢异喹啉类生物碱jorumycin的结构复杂及其高抗癌抗肿瘤的生物活性,有机化学家对它产生了浓厚的兴趣,纷纷进行了全合成研究。本文对该化合物的全合成研究进展进行了综述。
2 天然产物(-)-jorumycin的合成研究
2.1 Williams小组的全合成报道
2005年,科罗拉多州立大学的Williams小组[5]以多取代苯甲醇1为原料,通过手性吗啉酮和苄碘的衍生物立体选择性烷基化反应为关键步骤合成1个关键中间体2。从该中间体出发,经6步反应合成手性氨基酸3,经10步转化得四氢异喹啉4,然后通过肽缩合将氨基酸3与片段4偶联起来生成中间体5,接着经甲酸掉TBS、Swern氧化和甲磺酸催化分子内的Pictet-Spengler反应构建C,D环得到化合物6。通过研究他们发现化合物6中C3位上的构型与天然产物的相反,原因可能是由于在关环过程中的C3位发生了构型反转造成的。化合物6经5步反应得到了与天然产物(-)-jorumycin在C3位上构型相反的差向异构体3-epi-jorumycin。 见图1。
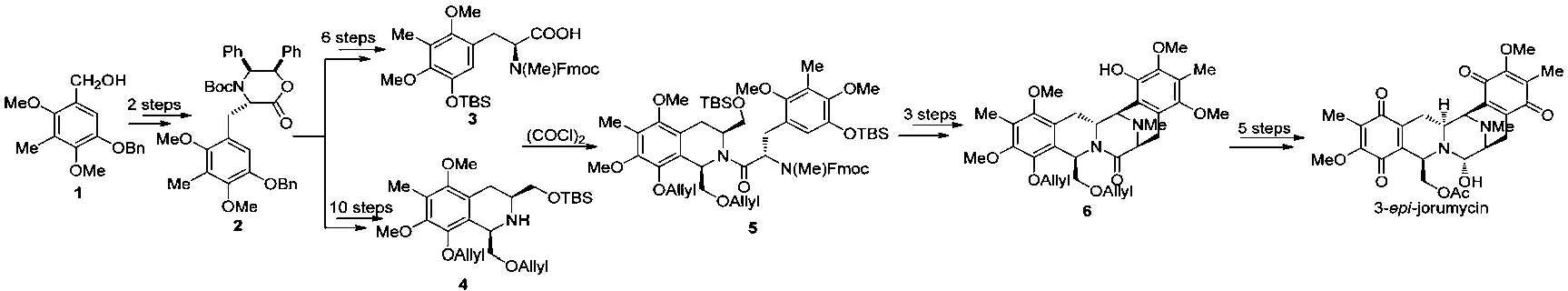
图1 合成结构示意图
随后,他们通过改变反应策略,首次全合成出目标化合物(-)-jorumycin。从化合物2出发,经5步反应合成氨基酸7,通过酰胺化反应使化合物4和氨基酸7偶联在一起生成中间体8。接着通过保护基的变化、氧化等5步反应得化合物9,再经分子内Pictet-Spengler反应、还原甲基化反应得五环中间体10。化合物10再经5步反应得到目标化合物(-)-jorumycin。该路线以多取代苄醇1计,共经历25步反应,总收率为7.2%,构环顺序为B→C,D。见图2。
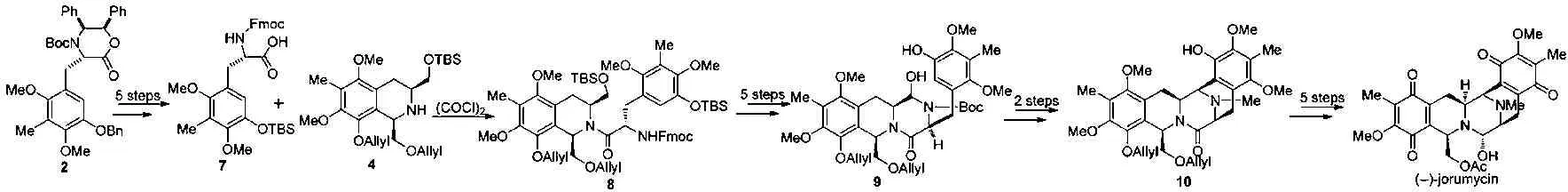
图2 全合成示意图
2.2 祝介平小组的全合成报道
2009年,法国国家科学研究中心的祝介平课题组[6]报道了(-)-jorumycin全合成研究。以丝氨酸衍生物11为原料,经4步和5步反应分别合成手性氮杂环丙烷衍生物12和13。从2,6-二甲氧基-甲苯14出发,经4步反应制得芳基溴化物15。然后将化合物15转化为镁格氏试剂后与环氮化合物12在Knochel发展的条件下发生反应生成四取代的苯丙氨酸衍生物16,接着掉O-Boc后与醛13发生分子间的Pictet-Spengler反应顺利构建D环生成中间体17。用苄基保护化合物17中的游离氨基后,再与另一分子的镁格氏试剂在Knochel's条件下发生反应,接着与苄氧基乙醛发生分子间的Pictet-Spengler反应构建出B环得中间体18,经还原、氧化和Strecker反应关上C环得五环中间体19。化合物19经掉苄基、氧化A、E环成双醌得天然产物(-)-jorunnamycin A,再经2步转化合成出目标化合物(-)-jorumycin。该路线新颖之处在于关环顺序变为D→B→C,应用了两次芳基镁试剂与环氮化合物在Knochel's条件下发生反应、2次Pictet-Spengler反应、1次Strecker反应构建出主体5环骨架,路线较以前更为简短。从化合物11出发,经18步,5.8%的总收率得(-)-jorumycin。见图3。
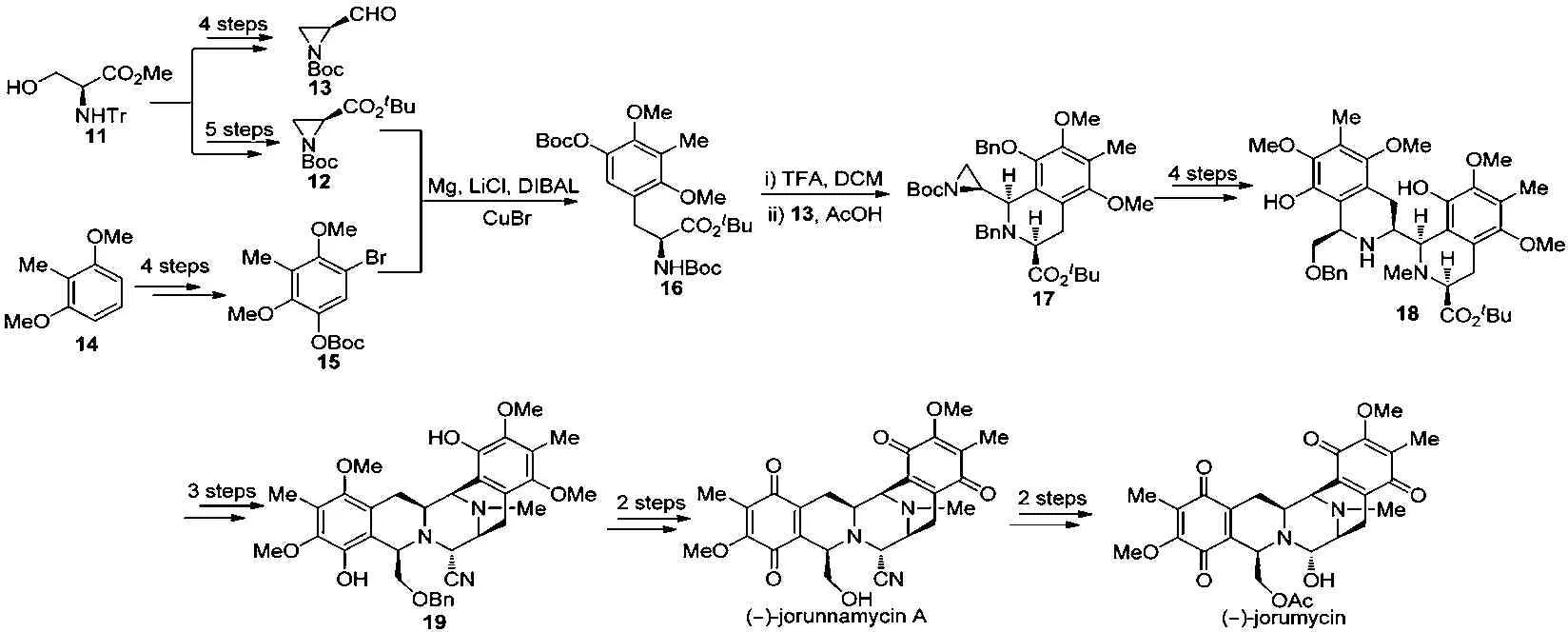
图3 全合成示意图
2.3 刘站柱课题组的合成报道
2012年,中国医学科学院北京协和医学院刘站柱课题[7]也报道了(-)-jorumycin的合成研究,他们从合成(-)-renieramycin G时[9]的一个关键中间体20出发,经四氢铝锂还原酰胺基、Strecker反应、氧化成醌、酯化反应等5步反应合成了(-)-jorumycin。见图4。

图4 全合成示意图
2.4 陈小川课题组的全合成报道
2013年,我们课题组也对天然产物(-)-jorumycin进行了全合成研究报道[9]。发展了一条使用2个源自L-酪氨酸的片段醇22和醛23,经高选择性的Pictet-Spengler环化偶联反应来合成(-)-jorumycin的新路线。从廉价易得的天然手性源L-酪氨酸出发,经8步反应高产率地得到三取代苯丙氨醇22[10]。化合物22经分子间的Pictet-Spengler反应、氨基保护、氧化等4步反应合成醛23,接着与氨基醇22再通过一次分子间的Pictet-Spengler反应高产率、高选择性地生成双四氢异喹啉24。然后化合物24经氨基、酚羟基保护、氧化和Strecker反应关上C环得五环化合物25,再经脱保护基、还原甲基化和掉苄基等3步反应得到中间体26。氧化化合物26的 A、E环成双醌得天然产物(-)-jorunnamycin A,再经2步转化合成出目标化合物(-)-jorumycin。见图5。
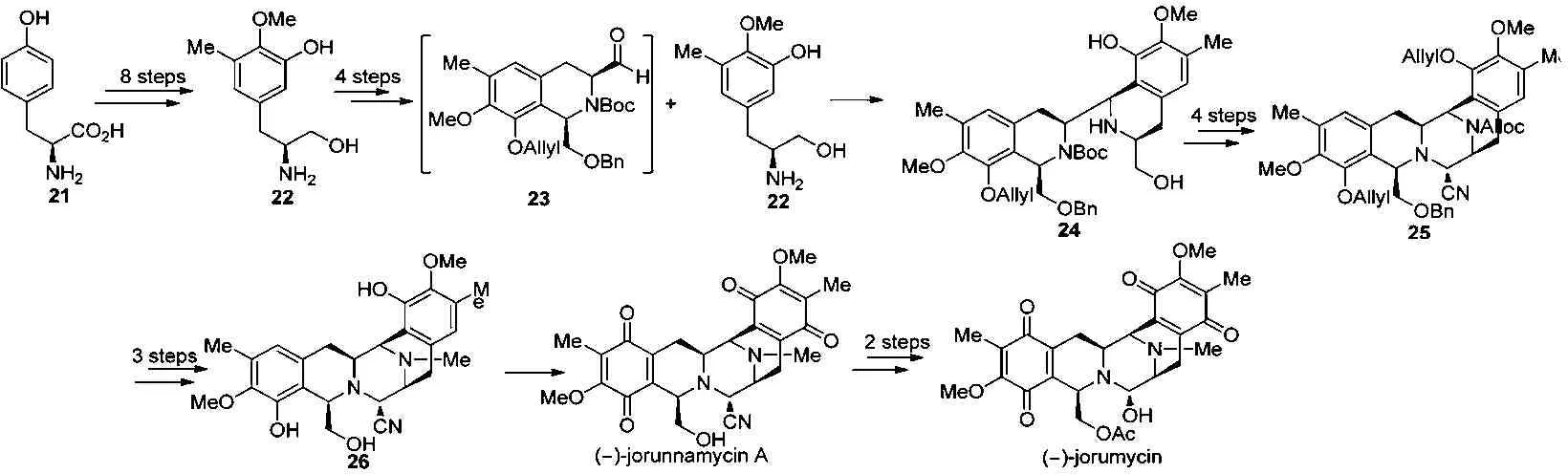
图5 全合成示意图
我们的合成策略是以L-酪氨酸为原料,经23步,14.1%的总收率合成了(-)-jorumycin。此合成方法新颖之处在于路线中2个偶联片段均由L-酪氨酸用类似的方法制得,不需再额外合成任何其它子片段。与以前的路线相比,经高选择性的Pictet-Spengler反应将左半部分片段醛和右半部分氨基醇偶联在一起,构建出D环,最后通过分子内的Strecker反应构建出C环,关环顺序是B→D→C。操作简单,原料便宜,并且可以放大生产,也是目前为止总产率最高的一条路线。
3 总结与展望
具有抗肿瘤活性的四氢异喹啉类生物碱从第1例具有抗癌活性的naphthriydinomycin被分离出来至今,已经被研究了近40年,期间一直有大量具有良好抗癌活性的分子被分离鉴定。鉴于四氢异喹啉类生物碱的结构多样性及其高抗癌抗肿瘤的生物活性,有机化学家对它们产生了浓厚的兴趣,由于大多数这类生物碱在天然生物中含量低,来源有限,限制了对其大规模的活性筛选,所以其化学合成研究就显得十分重要和紧迫。而在对这些具有抗肿瘤活性的四氢异喹啉类生物碱进行全合成的过程中,能够应用已有的合成方法学并检验其适用性及局限性。因此,无论从基础研究还是从应用研究的角度出发,对具有高抗癌生物活性的四氢异喹啉类生物碱进行全合成研究都具有重要的现实意义。
[1] Scott JD,Williams RM.Chemistry and Biology of the Tetrahydroisoquinoline Antitumor Antibiotics[J].Chem Rev,2002,102,1669-1730.
[2] Kluepfel D,Baker HA,Piattoni G,et al .Naphthyridinomycin,a new broad-spectrum antibiotic[J].J Antibiot,1975,28(7):497-502.
[3] Fontana A,Cavaliere P,Wahidulla S,et al.A New Antitumor Isoquinoline Alkaloid from the Marine Nudibranch Jorunna funebris[J].Tetrahedron,2000,56(37):7305-7308.
[4] Saito N,Tanaka C,Koizumi Y,et al.Chemistry of renieramycins.Part 6:Transformation of renieramycin M into jorumycin and renieramycin J including oxidative degradation products,mimosamycin,renierone,and renierol acetate[J].Tetrahedron,2004,60(17):3873-3881.
[5] Lane JW,Chen Y,Williams RM.Asymmetric Total Syntheses of(-)-Jorumycin,(-)-Renieramycin G,3-epi-Jorumycin,and 3-epi-Renieramycin G[J].J Am Chem Soc,2005,127(36):12684-12690.
[6] Wu YC,Zhu J.Asymmetric Total Syntheses of(-)-Renieramycin M and G and(-)-Jorumycin Using Aziridine as a Lynchpin[J].Org Lett,2009,11(23):5558-5561.
[7] Liu W,Liao X,Dong W,et al.Total synthesis and cytotoxicity of(-)-jorumycin and its analogues[J].Tetrahedron,2012,68(13):2759-2764.
[8] Liao XW,Liu W,Dong WF,et al .Total synthesis of(-)-renieramycin G from L-tyrosine[J].Tetrahedron,2009,65(29-30):5709-5715.
[9] Chen R,Liu H,Chen X.Asymmetric Total Synthesis of(-)-Jorunnamycin A and C and(-)-Jorumycin from L-Tyrosine[J].J Nat Prod,2013,76(9):1789-1795.
[10] Chen R,Zhu D,Hu Z,et al.A new approach to the synthesis of L-3-hydroxy-4-methoxy-5-methyl-phenylalanine derivatives from L-tyrosine[J].Tetrahedron:Asymmetry,2010,21(1):39-42.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
中成药(2017年12期)2018-01-19
中成药(2017年7期)2017-11-22
中成药(2017年8期)2017-11-22
国外医药(抗生素分册)(2016年1期)2016-07-10
合成化学(2015年1期)2016-01-17
股市动态分析(2015年12期)2015-09-10
中国当代医药(2015年24期)2015-03-01
安徽工业大学学报(自然科学版)(2014年4期)2014-07-11
