
三维有序结构In掺杂TiO2薄膜的可见光催化活性
2014-09-17 06:59王景声王恩君于彦龙郭丽梅曹亚安
物理化学学报 2014年3期
王景声 王恩君 于彦龙 郭丽梅 曹亚安,*
(1南开大学物理科学学院,天津300071;2南开大学泰达应用物理研究院,弱光非线性光子学教育部重点实验室,天津300457;3中国科学院合肥物质科学研究院,合肥230031)
1 引言
TiO2具有光催化效率高和化学稳定性强等特点,多年来在光催化研究领域倍受重视.1-3然而,TiO2的禁带宽度是3.2 eV,仅吸收紫外光(λ>387.5 nm),而对可见光无吸收(太阳光谱中紫外光部分仅占8%,而可见光占45%以上),从而导致了太阳能利用率的降低.另外,TiO2光生载流子的复合导致了光催化效率的降低.
金属或非金属掺杂是提高TiO2催化剂可见光催化活性的有效方法.Wang等4制备了Fe掺杂的TiO2催化剂,其光催化活性高于纯TiO2.Jing、5Fresno6和Yu7等对Sn掺杂TiO2进行了研究,均发现其光催化活性有明显提高.此外,TiO2掺杂La3+、Pd2+、Cr3+、Ag+以及稀土金属离子(Sm3+、Nd3+、Pr3+)等也见报道.8-12近年来Cao等报道了Sn、13Zr、14Ni、15In、16N17和B18等离子掺杂TiO2,结果表明,金属或非金属离子掺杂增强了TiO2的可见光响应,促进了光生载流子的分离,有效地提高了催化剂的可见光催化活性.
三维有序结构TiO2薄膜具有比表面积大、吸附能力强和光的利用率高等特点,表现出优异的光催化活性,近年来已成为光催化领域研究的热点.19-22King等23采用低温原子层沉积的方法制备了反蛋白石结构TiO2;Ren等24用模板法制备了具有光子禁带的反蛋白石结构的TiO2,紫外光催化结果表明,该催化剂光子效率比P25高248%;Doong等25制备了高度有序的反蛋白石结构的TiO2,其孔径范围在480-1000 nm之间,结果表明,该催化剂的紫外光催化的降解率随着孔径的增大而线性提高;Li和Shang26制备了N掺杂反蛋白石结构TiO2,研究发现该催化剂在可见光区吸收增强、光催化活性明显提高.
直至目前,三维有序结构In离子掺杂的TiO2薄膜可见光催化剂未见报道;对于In离子的掺杂方式和掺杂能级等掺杂机理尚不清楚;In离子掺杂对催化剂光生载流子过程和光催化活性的影响,以及可见光催化机理还有待于研究.
本文采用自组装生长聚苯乙烯胶体模板方法和溶胶-凝胶法制备出三维有序结构In掺杂TiO2薄膜可见光催化剂.与TiO2和三维有序结构TiO2薄膜相比,该催化剂的可见光催化活性显著提高.利用X射线电子衍射(XRD)谱、X射线光电子能谱(XPS)和紫外-可见漫反射吸收光谱(UV-Vis DRS)等表征技术,确定了催化剂的结构、In离子的掺杂方式和能带结构,并分析讨论了三维有序结构In掺杂TiO2薄膜降解甲醛的可见光催化机理.
2 实验部分
2.1 样品的制备
将洗净的载玻片垂直放入盛有聚苯乙烯微球的悬浊液中(悬浊液的体积为8.5 mL,聚苯乙烯微球的体积分数为0.2%),在干燥箱中55°C恒温沉化,待溶剂完全蒸发后,载玻片表面生长出聚苯乙烯微球胶体薄膜,然后在80°C加热2 h,得到聚苯乙烯微球胶体模板.
室温下将 4.4 mL 的 InCl3溶液(0.6 mol·L-1)与40 mL无水乙醇混合,在剧烈搅拌下,将12 mL的Ti(OC4H9)4缓慢滴加到上述混合液中,然后再加入一定量的盐酸(12 mol·L-1),调节pH为3.5,经剧烈搅拌后,得到稳定透明的TiO2溶胶.
将聚苯乙烯微球胶体模板垂直浸泡在TiO2溶胶中(约1 min),以2.5 mm·s-1的速率垂直提拉成膜,空气中自然干燥,上述过程重复两次.将模板移至马弗炉中,以5°C·min-1的速率缓慢升温至500°C,焙烧2 h后自然冷却,制备出三维有序结构In掺杂的TiO2(IO-TiO2-In)薄膜,放入干燥器备用.
三维有序结构TiO2(IO-TiO2)薄膜制备方法同上,区别为TiO2溶胶中未加入InCl3.采用垂直提拉法制备出TiO2溶胶薄膜,500°C焙烧2 h(升温速率为5 °C·min-1),制得纯TiO2薄膜.
实验中所用试剂均为分析纯,实验用水为ρ≥18.0 MΩ·cm高纯水.
2.2 物性表征
X射线衍射(XRD,Rigaku D/max-2500,Cu靶,Kα线)测定了薄膜样品的晶体结构;X射线光电子能谱(XPS)的测试在ESCA Lab 250型X光电子能谱仪(Mg-Al靶,Kα线)上进行,所有的谱图依据C 1s(284.6 eV)进行校正;利用扫描电子显微镜(SEM,JEOL JSM-6700F)对薄膜样品的表面形貌和厚度进行观测;利用紫外-可见分光光度计(U-4100,Hitachi)测定薄膜样品的紫外可见漫反射吸收光谱.
2.3 光催化反应
光催化降解甲醛的实验在密封的Pyrex玻璃反应器(455 mL)中进行,以500 W的Xe灯作为催化反应的外照光源,辐射波长>290 nm(在可见光催化实验中,反应器前放置400 nm的截止滤光片);薄膜催化剂平铺在反应器底部(面积为2 cm×4 cm),膜面朝光,并与光照方向保持垂直,光源距反应器10 cm;反应器内加入15 μL甲醛(优级纯99.9%),在干燥箱中60°C恒温40 min,使甲醛充分挥发形成气体;反应器与光源之间放置隔热玻璃,恒温保持反应体系的温度((30±2)°C)与浓度(1.19×10-3mol·L-1)的平衡.利用气相色谱仪(GC-7890F,天美)每隔80 min(紫外光40 min)监测甲醛光催化降解生成CO2的浓度变化;色谱柱为TDX-01(1 m×ϕ 3 mm),高纯氮气(99.99%)为载气,在氢火焰检测器与色谱柱间装有转化炉,使CO2发生加氢反应(CO2+4H2→CH4+2H2O),从而测定CO2的浓度.
3 结果与讨论
3.1 光催化活性
采用光催化降解甲醛生成CO2(最终产物)的浓度来评价催化剂的光催化活性.图1为TiO2、IO-TiO2和IO-TiO2-In薄膜可见光催化降解甲醛生成CO2的曲线,其可见光催化活性见表1.结果表明,在可见光光解实验中,光照160 min仅有微量的CO2产生(2.99×10-9mol·L-1);与光解实验相比,TiO2薄膜几乎无可见光活性(CO2浓度为2.99×10-9mol·L-1);IOTiO2薄膜的可见光活性(CO2浓度为4.01×10-9mol·L-1,CO2生成率为 8.55×10-11mol·cm-2·h-1)高于TiO2;然而,IO-TiO2-In薄膜表现出最高可见光活性(CO2浓度为1.89×10-8mol·L-1,CO2生成率为4.03×10-10mol·cm-2·h-1),其产生 CO2浓度和 CO2生成率大约是TiO2和IO-TiO2的5倍.图2为TiO2、IO-TiO2和IO-TiO2-In薄膜紫外光催化降解甲醛生成CO2的曲线,其紫外光催化活性见表1.样品的紫外光催化活性遵循IO-TiO2-In>IO-TiO2>TiO2规律,与可见光催化相似.
3.2 催化剂的性质
图3为TiO2、IO-TiO2和IO-TiO2-In薄膜的SEM照片.结果表明,TiO2薄膜(图3a)是由堆积致密的TiO2纳米粒子组成,其粒径约为20-40 nm.IO-TiO2薄膜(图3b)呈现三维周期有序的多层多孔结构,每层由六边形骨架连接而成,相邻六边形中心间距约为200 nm.IO-TiO2-In薄膜(图3c)的结构与IO-TiO2相同.

图1 TiO2(b)、IO-TiO2(c)和IO-TiO2-In(d)薄膜可见光降解甲醛生成CO2的曲线Fig.1 CO2generation curves of photocatalytic degradation of formaldehyde under visible light with TiO2(b),IO-TiO2(c),and IO-TiO2-In(d)films(a)Blank is the photolysis of HCHO.

图2 TiO2(b)、IO-TiO2(c)和IO-TiO2-In(d)薄膜紫外光降解甲醛生成CO2的曲线Fig.2 CO2generation curves of photocatalytic degradation of formaldehyde under UV light with TiO2(b),IO-TiO2(c),and IO-TiO2-In(d)films(a)Blank is the photolysis of HCHO.

表1 TiO2、IO-TiO2和IO-TiO2-In薄膜可见和紫外光催化活性Table 1 Photocatalytic activity of TiO2,IO-TiO2,and IO-TiO2-In films under visible and UV light irradiation

图3 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的扫描电子显微镜(SEM)照片Fig.3 Scanning electron microscopy(SEM)images of TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
图4 为TiO2、IO-TiO2和IO-TiO2-In薄膜的XRD谱.在25.2°、37.8°、48.0°、53.8°和 54.9°附近的衍射峰分别对应于锐钛矿 TiO2(101)、(004)、(200)、(105)和(211)晶面,27表明所有样品主要为四方晶系锐钛矿结构.在图4(c)中,22.3°、30.9°、45.5°和58.2°附近出现的衍射峰符合In2O3的衍射峰(ICSD 21-1272),表明在掺杂过程中In离子在TiO2表面可能形成In2O3物种.以上结果能够由XPS谱进一步证明.图5为TiO2、IO-TiO2和 IO-TiO2-In薄膜的Ti 2p的 XPS谱.结合能为458.4和464.2 eV的峰分别为锐钛矿TiO2的晶格 Ti的 Ti 2p3/2和 Ti 2p1/2;16而在 TiO2、IOTiO2和IO-TiO2-In薄膜的O 1s的XPS谱(图6)中,结合能为529.6、530.7和532.0 eV的峰(XPSPEAK41软件拟合)分别归属为锐钛矿TiO2的晶格氧、表面桥氧和表面羟基(OH)氧,28由此证明TiO2、IO-TiO2和IO-TiO2-In薄膜样品均为锐钛矿结构.,
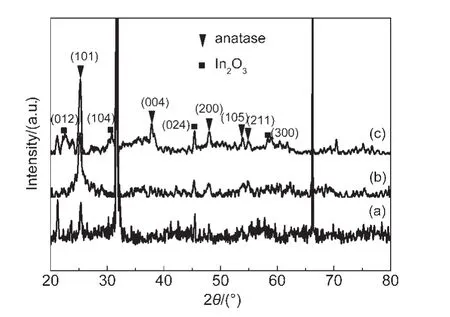
图4 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的X射线衍射(XRD)谱Fig.4 X-ray diffraction(XRD)spectra of TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films

图5 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的Ti 2p的X射线光电子能谱(XPS)图Fig.5 Ti 2p X-ray photoelectron spectroscopies(XPS)for TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films

图6 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的O 1s的XPS图Fig.6 O 1s XPS for TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films

图7 IO-TiO2-In薄膜In 3d的XPS谱Fig.7 In 3d XPS for IO-TiO2-In films
图7 为IO-TiO2-In薄膜In 3d的XPS谱.拟合后,在结合能为444.6和452.1 eV的一对强峰与In2O3中In 3d5/2和In 3d3/2的结合能一致,29表明掺入的In离子主要是以In2O3物种的形式存在于IO-TiO2-In薄膜表面,这与XRD表征结果一致.另外,基于XPS原理分析,由于In的电负性(1.7)大于Ti的电负性(1.5),如果In离子进入TiO2的晶格,取代并占据Ti离子的晶格位置,将会导致TiO2中晶格Ti和晶格O的结合能提高.16然而,与TiO2比较,IO-TiO2-In薄膜的Ti 2p和O 1s谱(图5和图6)中的晶格Ti 2p和晶格O 1s的峰未发生明显高位移动,进一步表明掺杂的In离子没有进入TiO2晶格,而是作为In2O3物种存在于TiO2表面,形成TiO2与In2O3复合结构(TiO2/In2O3).

图8 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的Cl 2p的XPS图Fig.8 Cl 2p XPS for TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
然而,在拟合后的In 3d谱中,445.3和452.9 eV的一对弱峰,其In 3d5/2的结合能(445.3 eV)位于In2O3中In 3d5/2的结合能(444.6 eV)30和 InCl3中 In 3d5/2的结合能(446.0 eV)31之间,意味着掺入的In离子可能与O和Cl离子连接.对于IO-TiO2-In薄膜Cl 2p3/2谱(图8c),Cl 2p3/2的结合能(198.64 eV)高于TiCl4中Cl 2p3/2的结合能(198.20 eV),而低于InCl3中Cl 2p3/2的结合能(199.1 eV),31表明Cl离子可能与In连接.然而Cl的离子半径(0.181 nm)大于O离子半径(0.140 nm),不可能进入TiO2晶格取代晶格O,Cl仅能存在于样品表面.32基于以上分析,在IO-TiO2-In薄膜中In离子的掺杂态能够被确定:In离子分别与表面的O和Cl离子连接,在IO-TiO2-In薄膜表面形成O-In-Clx(x=1,2)物种.对于TiO2和IO-TiO2薄膜(图8(a,b)),Cl 2p3/2峰的结合能分别为198.36和198.39 eV,与TiCl4的Cl 2p3/2结合能(198.2 eV)33接近,表明 Cl−离子能够与Ti4+连接,在TiO2和IO-TiO2表面形成Ti-Clx(x=1,2)物种.
图9为TiO2、IO-TiO2和IO-TiO2-In薄膜的紫外-可见光漫反射吸收谱.在小于400 nm的强峰为TiO2的带-带跃迁.起峰阈值分别为374.7、374.9和382.5 nm,相应的禁带宽度为3.37、3.33和3.12 eV.另外,IO-TiO2-In样品在375-480 nm出现的肩峰为表面In2O3物种的价带到导带的跃迁(In2O3的禁带宽度为2.8 eV34);而500-800 nm吸收峰归属为价带到O-In-Clx物种的表面态能级的跃迁,表面态能级约在导带下0.5-1.6 eV范围.16以上结果表明,In离子的掺杂在IO-TiO2-In薄膜的表面形成In2O3和O-In-Clx物种,既增强了IO-TiO2-In薄膜的可见光吸收,又有利于可见光催化活性的提高.
3.3 光催化机理

图9 TiO2(a)、IO-TiO2(b)和IO-TiO2-In(c)薄膜的紫外-可见光漫反射吸收光谱Fig.9 UV-Vis diffuse reflectance spectra of TiO2(a),IO-TiO2(b),and IO-TiO2-In(c)films
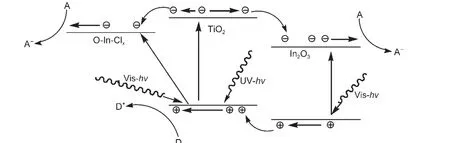
图10 IO-TiO2-In薄膜的能带结构及可见和紫外光催化机理示意图Fig.10 Schematic diagram of energy band structures and photocatalytic mechanism under visible and UV light for IO-TiO2-In film
依据以上表征结果,IO-TiO2-In薄膜的能带结构见图10,其可见、紫外光催化机理如下.对于可见光催化,TiO2和IO-TiO2薄膜可见光活性低的主要原因是TiO2和IO-TiO2的禁带宽度(3.20 eV)较大,对于可见光(λ>400 nm)没有吸收.与TiO2和IO-TiO2薄膜相比,IO-TiO2-In薄膜具有较高的可见光活性主要归因于In离子掺杂.In离子掺杂在IO-TiO2-In薄膜表面主要形成In2O3和O-In-Clx(x=1,2)物种;In2O3的禁带宽度为2.8 eV,34可以吸收可见光,提高了可见光利用率;而TiO2和In2O3的复合有效地提高了光生载流子的分离(阻止其复合).35另外,O-In-Clx(x=1,2)物种的表面态能级在导带下0.5-1.6 eV范围,在可见光(λ>400 nm)的照射下,催化剂产生价带到表面态能级的跃迁,增加了可见光的吸收,导致了光生载流子的直接分离.由In2O3的带-带跃迁和TiO2导带到O-In-Clx表面态能级跃迁产生的光生电子能够被表面吸附的O2分子俘获形成O-2,进一步氧化表面吸附甲醛分子.In2O3价带的光生空穴经TiO2/In2O3界面,与TiO2价带的光生空穴一起直接氧化表面吸附甲醛分子,导致了IO-TiO2-In薄膜催化剂可见光催化活性的有效提高.
对于紫外光催化,IO-TiO2-In薄膜相对TiO2和IO-TiO2薄膜紫外光活性进一步提高的原因主要决定于催化剂的能带结构(图10).TiO2和IO-TiO2薄膜具有较高的紫外光活性归因于带-带跃迁的光生电子和光生空穴在催化剂表面的氧化还原作用;36然而,导带大部分的光生电子在转移到表面过程中由无辐射跃迁到氧空位能级与价带的光生空穴复合,仅有少数的光生电子和光生空穴到达催化剂的表面参加表面甲醛分子降解反应.对于IO-TiO2-In薄膜,TiO2带-带跃迁的光生电子可以转移到O-In-Clx(x=1,2)物种的表面态能级或转移In2O3导带到达催化剂表面;In2O3价带的光生空穴能够转移到TiO2价带,与TiO2价带的光生空穴一道到达催化剂的表面.与TiO2和IO-TiO2薄膜相比,以上电荷转移过程有效地提高了光生载流子的分离效率,阻止光生载流子复合,能够使更多的光生电子和光生空穴到达催化剂的表面参加表面甲醛分子降解反应,导致了催化剂紫外光催化活性的有效提高.
另外,IO-TiO2和IO-TiO2-In薄膜具有三维有序结构,与TiO2薄膜相比,比表面积大,光的利用率高,更有利于紫外和可见光催化活性的提高.
4 结论
采用自组装生长聚苯乙烯胶体模板法和溶胶-凝胶法制备出高活性的三维有序结构In掺杂TiO2薄膜可见光催化剂(IO-TiO2-In).与TiO2相比,IOTiO2-In薄膜不但比表面积大,光的利用率高,而且表面形成In2O3和O-In-Clx(x=1,2)物种,即增强了可见光的吸收,又促进了光生载流子的分离,有效地提高了光生电子和光生空穴在固/气界面参加光催化反应的利用率,使IO-TiO2-In薄膜催化剂的可见光催化活性显著提高.
(1)Choi,W.;Termin,A.;Hoffmann,M.R.J.Phys.Chem.1994,98,13669.doi:10.1021/j100102a038
(2) Ghicov,A.;Macak,J.M.;Tsuchiya,H.;Kunze,J.;Haeublein,V.;Frey,L.;Schmuki,P.Nano Lett.2006,6,1080.doi:10.1021/nl0600979
(3)Chen,X.B.;Mao,S.S.Chem.Rev.2007,107,2891.doi:10.1021/cr0500535
(4)Wang,C.;Bahnemanna,D.W.;Dohrmannb,J.K.Chem.Commun.2000,1539.
(5) Jing,L.Q.;Fu,H.G.;Wang,B.Q.;Wang,D.J.;Xin,B.F.;Li,S.D.;Sun,J.Z.Appl.Catal.B 2006,62,282.doi:10.1016/j.apcatb.2005.08.012
(6) Fresno,F.;Tudela,D.;Coronado,J.M.;Fernández-Gracía,M.;Hungría,A.B.;Soria,J.Phys.Chem.Chem.Phys.2006,8,2421.doi:10.1039/b601920j
(7)Yu,J.G.;Liu,S.W.;Zhou,M.H.J.Phys.Chem.C 2008,112,2050.doi:10.1021/jp0770007
(8) Huo,Y.N.;Zhu,J.;Li,J.X.;Li,G.S.;Li,H.X.Journal of Molecular Catalysis A:Chemical 2007,278,237.doi:10.1016/j.molcata.2007.07.054
(9) Wu,C.;Chao,C.;Kuo,F.Catal.Today 2004,97,103.doi:10.1016/j.cattod.2004.04.055
(10) Anpo,M.;Takeuchi,M.J.Catal.2003,216,505.doi:10.1016/S0021-9517(02)00104-5
(11)Wang,P.;Wang,D.J.;Xie,T.F.;Li,H.Y.;Yang,M.;Wei,X.Mater.Chem.Phys.2008,109,181.doi:10.1016/j.matchemphys.2007.11.019
(12) Liang,C.H.;Li,F.B.;Liu,C.S.;Lu,J.L.;Wang,X.G.Dyes and Pigments 2008,76,477.doi:10.1016/j.dyepig.2006.10.006
(13)Cao,Y.Q.;He,T.;Zhao,L.S.;Wang,E.J.;Yang,W.S.;Cao,Y.A.J.Phys.Chem.C 2009,113,18121.doi:10.1021/jp9069288
(14)Wang,E.J.;Yang,H.Y.;Cao,Y.A.J.Chem.2009,67,2759.
(15) Luo,D.C.;Zhang,L.L.;Long,H.J.;Chen,Y.M.;Cao,Y.A.Acta Phys.-Chim.Sin.2008,24,1095.[罗大超,张兰兰,龙绘锦,陈咏梅,曹亚安.物理化学学报,2008,24,1095.]doi:10.3866/PKU.WHXB20080632
(16)Wang,E.J.;Yang,W.S.;Cao,Y.A.J.Phys.Chem.C 2009,113,20912.doi:10.1021/jp9041793
(17) Cao,Y.Q.;He,T.;Chen,Y.M.J.Phys.Chem.C 2010,114,3627.doi:10.1021/jp100786x
(18)Yuan,J.X.;Wang,E.J.;Chen,Y.M.;Yang,W.S.;Yao,J.H.;Cao,Y.A.Appl.Surf.Sci.2011,257,7335.doi:10.1016/j.apsusc.2011.03.139
(19) Chen,J.I.L.;Freymann,G.;Choi,S.Y.;Kitaev,V.G.;Ozin,A.Adv.Mater.2006,18,1915.
(20) Chen,I.L.;Freymann,G.V.;Kitaev,V.;Ozin,G.A.J.Am.Chem.Soc.2007,129,1196.doi:10.1021/ja066102s
(21) Chen,J.I.L.;Loso,E.;Ebrahim,N.;Ozin,G.A.J.Am.Chem.Soc.2008,130,5420.doi:10.1021/ja800288f
(22) Chen,J.I.L.;Freymann,G.;Choi,S.Y.;Kitaev,V.;Ozin,G.A.J.Mater.Chem.2008,18,369.doi:10.1039/b708474a
(23) King,J.S.;Graugnard,E.;Summers,C.J.Adv.Mater.2005,17,1010.
(24) Ren,M.;Ravikrishna,R.;Valsaraj,K.T.Environ.Sci.Technol.2006,40,7029.doi:10.1021/es061045o
(25) Doong,R.A.;Chang,S.M.;Hung,Y.C.Sep.Purif.Technol.2007,58,192.doi:10.1016/j.seppur.2007.07.029
(26) Li,Q.;Shang,J.K.J.Am.Chem.Soc.2008,91,660.
(27) Gao,B.F.;Ma,Y.;Cao,Y.A.;Yang,W.S.;Yao,J.N.J.Phys.Chem.B 2006,110,14391.doi:10.1021/jp0624606
(28)Cao,Y.A.;Yang,W.S.;Chen,Y.M.;Du,H.;Yue,P.Appl.Surf.Sci.2004,236,223.doi:10.1016/j.apsusc.2004.04.020
(29) Li,J.;Zeng,H.C.J.Am.Chem.Soc.2007,129,5839.
(30) Reddya,B.M.;Chowdhury,B.;Smirniotis,P.G.Appl.Catal.A 2001,219,53.doi:10.1016/S0926-860X(01)00658-5
(31) Freeland,B.H.;Habeeb,J.J.;Tuck,D.G.Can.J.Chem.1977,55,1527.doi:10.1139/v77-213
(32) Zhu,J.;Zheng,W.;He,B.;Zhang,J.L.;Anpob,M.J.Mol.Catal.A:Chem.2004,216,35.doi:10.1016/j.molcata.2004.01.008
(33) Mousty-Desbuquoit,C.;Riga,J.;Verbist,J.J.J.Chem.Phys.1983,79,26.doi:10.1063/1.445567
(34) Poznyak,S.K.;Talapin,D.V.;Kulak,A.I.J.Phys.Chem.B 2001,105,4816.doi:10.1021/jp003247r
(35) Cao,Y.A.;Zhang,X.T.;Yang,W.S.;Du,H.;Bai,Y.B.;Li,T.J.;Yao,J.N.Chem.Mater.2000,12,3445.doi:10.1021/cm0004432
(36) Long,H.J.;Wang,E.J.;Dong,J.Z.;Wang,L.L.;Cao,Y.Q.;Yang,W.S.;Cao,Y.A.J.Chem.2009,67,1533.
猜你喜欢
科学之友(2022年11期)2022-11-03
大学物理(2022年9期)2022-09-28
工业水处理(2022年6期)2022-06-23
石油化工高等学校学报(2021年3期)2021-07-15
物理通报(2020年7期)2020-07-01
四川冶金(2019年5期)2019-12-23
物理化学学报(2017年3期)2017-03-11
电线电缆(2016年5期)2016-02-27
原子与分子物理学报(2015年3期)2015-11-24
中国塑料(2014年1期)2014-10-17
