
噻吩在M(111)(M=Pd,Pt,Au)表面的吸附
2014-09-17 07:00:00张连阳夏盛杰倪哲明
物理化学学报 2014年12期
施 炜 张连阳 夏盛杰 倪哲明
(浙江工业大学化学工程学院,先进催化材料实验室,杭州310032)
1 Introduction
Thiophene is one of the key components of fuel.The sulfurcontaining compounds which are produced by thiophene combustion can cause serious pollution to the environment.As environment protection has been a major concern of modern society,the sulfur content of fuel is now strictly regulated in every country.For this reason,the removal of sulfur from thiophene and other compounds has become the key point to produce low-sulfur fuel.1-4
At present,the most popular desulphurization methods include non-hydrogenation desulfurization and hydrodesulfurization.Compared to non-hydrodesulfurization,the hydrodesulfurization technology is more mature and the desulfurization is more significant.5Precious metal catalyst is a kind of common hydrogenation catalyst which has superior catalytic activity and more mild reaction conditions.It has been widely applied in the thiophene hydrodesulfurization reactions.6Among different surfaces of precious metal catalyst,the(111)surface is the most frequently studied in the hydrogenation of thiophene compared to the(100),7,8(110),9(0001)10surfaces.As it is the thermodynamically most stable surface that dominates in large particles in catalysts,it would lead to the higher selectivity and activity.Terada et al.11found that the S atom of thiophene interacted with Pd(111)surface and the transfer of electrons between Pd(111)surface and thiophene molecule led to a strong chemisorption.Sato et al.12have investigated the adsorption of thiophene and its compounds on the Pt(111)surface.They found that with the increasing of substituent groups in thiophene molecule,the adsorption energy and the desulfurization activity decreased gradually.Zhou et al.13have investigated the electronic structure of thiophene.They found that with the increasing of surface coverage,the adsorption of thiophene molecule decreased gradually.However,it is difficult to explain the adsorption process of thiophene on precious metal catalysts from experimental study,due to the complexity of experiment and the limitation of characterization methods.
Density functional theory(DFT)14,15can be used to calculate the adsorption energy,structure parameter,and charge population in the process of adsorption.Sitamraju et al.16have calculated the adsorption of thiophene with different functionals on TiO2surface.They found that the DFT+D method under GGA-PW91 functional was more accurate by using theoretical results compared with experimental values.Callsen et al.17have used ab initio method to investigate the adsorption mechanism of thiophene on Cu(111)surface.They found that the thiophene adsorbed on Cu(111)surface obliquely through the S atom and the adsorption belonged to the weak chemisorption.Wang et al.18have studied the optimal sturcture of thiophenic compounds adsorbed on different cationexchanged Y-zeolites.They found that the Cu(I)-zeolites showed the best capacity of adsorption desulfurization.We have calculated the hydrogenation mechanism of thiophene on Au(111)plane in our previous work.19We found that the reaction was most likely to occur on α-C and the hydroisomerization was identified as a primary mechanism.
To our knowledge,no theoretical investigations have been published for regarding the difference of thiophene on M(111)(M=Pd,Pt and Au)surfaces in detail.This motivates us to study the adsorption mechanism of thiophene on M(111)surfaces.In the present work,we use DFT method and Dmol3program package to calculate the adsorption of thiophene on the M(111)surfaces to further understand the difference between the adsorption of thiophene on Pd-,Pt-,andAu-based catalysts.Several adsorption geometries,including vertical and parallel,were explored in detail.Structure parameter,adsorption energy,charge population,and partial density of states(PDOSs)analyses were also carried out to discover the optimum geometric pattern of thiophene on M(111)surfaces.This work is devoted to provide the theory basis for the development of hydrodesulfurization catalyst about thiophene.
2 Methods and model construction
2.1 Surface model of M(111)
Selection of layers may influence on adsorption energy,but it will not impact the geometrical configuration of adsorbents and the stable conformation of adsorption site in accordance with previous studies.19-22In order to make a good compromise among the calculation accuracy,calculation efficiency,and the complexity of thiophene molecule,we modeled the M(111)surface by a periodic three-layer slab with a p(3×3)unit cell.There are four possible adsorption sites of thiophene,which includes top(0°,30°,60°),hcp(0°,30°,60°),fcc(0°,30°,60°),and bridge(0°,30°,60°)sites,as shown in Fig.1.The vacuum region thickness between the repeated slabs was 1 nm,which was large enough to avoid interactions between slabs.The layer and the vacuum region could form a unit which was repeated periodically in the space.Spin polarization was not considered in our calculations because its effect had been found to be negligible.19,21,23,24Only one thiophene molecule was adsorbed on one side of the slab,in other words,the coverage was 1/9 mono-layer(ML).In all geometry optimization calculations,the bottom layer for M(111)surface was kept fixed,while the rest of the atoms were allowed to relax freely.
2.2 Computational method
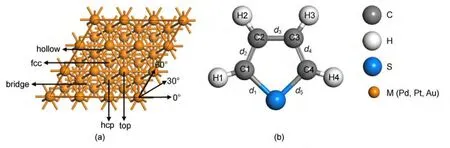
Fig.1 Top view of M(111)surface model(a)and the structure of thiophene(b)
All calculations were performed with the DMol3program package25in Materials Studio 5.5 of Accelrys Inc.The DFT slab approach with generalized gradient approximation of Perdew and Wang(GGA-PW91)26,27functional and a double numerical basis with polarization functions(DNP)were adopted.The Brillouin zone was sampled with the Monkhorst-Pack grid and the k-point was set to 3×3×1.28The Methfessel-Paxton smearing was 5×10-3Ha.The convergence criterion of optimal geometry was based on the energy,force,and displacement convergence,which were 2×10-5Ha,4×10-4Ha·nm-1,and 5×10-4nm,respectively.Under the present computational conditions,the lattice parameters of Pd(111),Pt(111),and Au(111)were calculated to be 0.3891,0.3924,0.4078 nm,respectively.Our results are in good agreement with the experimental values of 0.3895,0.3923,0.4080 nm,29,30respectively.These results showed that the method of calculation as given in this paper is reliable.
The adsorption energy can be used to describe the variation of total energy of each species before and after adsorption.The signs and values of adsorption energy can indicate the possibility of adsorption.In the present work,the adsorption energies(Eads)were calculated by:
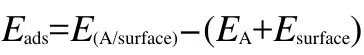
where E(A/surface)is the total energy of the thiophene molecule adsorption system in the equilibrium state on M(111)surface;EAand Esurfaceare the total energies of the free thiophene molecule and clean M(111)surface,respectively.With this definition,a negative value corresponds to the stable adsorption on M(111)surface.
3 Results and discussion
3.1 Adsorption energies and geometries analysis
According to the structure of thiophene and the result studied from reference,24there are two possible adsorption geometries.(1)Vertical adsorption:thiophene sits at the metal surface through sulfur atom on 12 different sits such as top,hcp,fcc,and bridge sites.(2)Parallel adsorption:thiophene sits at the metal surface through sulfur atom on thirty-six different adsorption sites,which included top(0°,30°,60°),hcp(0°,30°,60°),fcc(0°,30°,60°),and bridge(0°,30°,60°)sites.The 0°,30°,and 60°represent the adsorption sites with the symmetry axis rotating 0°,30°,and 60°from the horizontal direction,respectively.
Adsorption energies of thiophene molecule on M(111)surface are listed in the Table 1.It can be seen that:(1)in any case,the vertical adsorption geometry is less stable than the parallel adsorption geometry.These results are in good agreement with other authors′reports.24Out of our expectation,the adsorption energies of Pd-p-fcc-30°,Pt-v-hcp,and Pt-v-fcc sites are abnormal.This could be caused by the reason that the Pd-p-fcc-30°site was converted to vertical adsorption and the Pt-v-hcp,Pt-v-fcc sites were converted to parallel adsorption.In addition,the similar structure of thiophene after adsorption leads to the close adsorption energies.(2)There are differences in the adsorption energies of thiophene molecule on different metal surfaces,which follows the order of Pd,Pt,and Au.These results are in good agreement with the hydrodesulfurization activity of metals.31(3)For the adsorption of thiophene on Pd(111)surface,the p-fcc-0°site is the most stable adsorption and its Eadsis-163.7 kJ·mol-1,as shown in Fig.2(a);for the adsorption of thiophene on Pt(111)surface,the p-hcp-30°site is the most stable adsorption and its Eadsis-115.8 kJ·mol-1,as shown in Fig.2(b);and for the adsorption of thiophene on Au(111)surface,the p-fcc-30°site is the most stable adsorption and its Eadsis-55.9 kJ·mol-1,as shown in Fig.2(c).
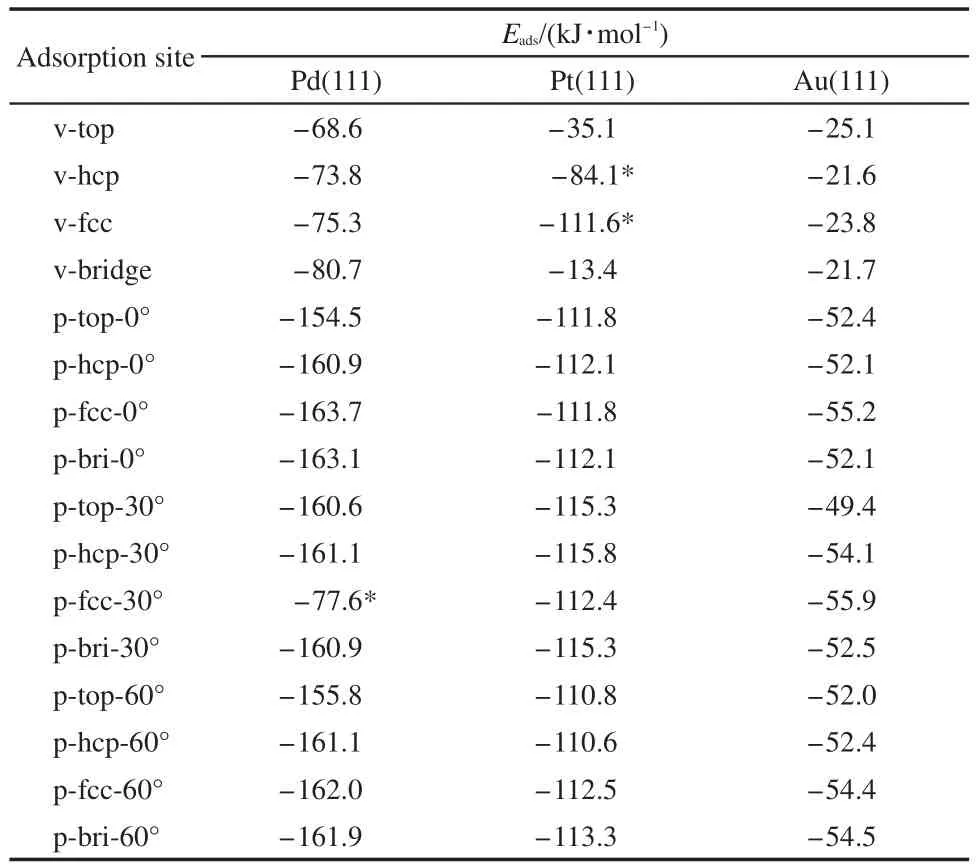
Table 1 Adsorption energies of thiophene molecule on M(111)surfaces
In order to analyze the structure parameter of thiophene adsorbed on different metal surfaces,Table 2 lists the adsorption energy and structure parameters of thiophene molecule on M(111)surface,Table 3 lists the distance between the atoms of thiophene and M(111)surface.By comparing Fig.2,Table 2,and Table 3,it can be seen that the calculation values of bond length for free thiophene are in good agreement with the experimental values.11,32After adsorption,all the bond lengths of thiophene increase,while the energy of thiophene decreases.These changes suggest that the adsorption of thiophene on M(111)surface could promote the hydrogenation reaction.
When thiophene molecule adsorbs on Pd-p-fcc-0°site,there was an elongation in the range of 0.0044-0.0124 nm for the bond lengths of d1
-d5(Fig.1).Both the bond lengths of d1and d5changed significantly,thed1was elongated from 0.1727 to 0.1851 nm and the d5was elongated from 0.1729 to 0.1853 nm.After the adsorption,structure relaxation of the top layer metal surface occurs and thiophene ring circles clockwise around the horizontal direction.The S atom of thiophene transfers to the hollow site between the fcc and top sites.The H atoms of thiophene move upward and the C2,C3 atoms(Fig.1)lie flush with the S atom.Finally,the thiophene adsorbs on the Pd(111)surface through the ring plane.
When thiophene molecule adsorbs on Pt-p-hcp-30°site,there is an elongation in the range of 0.0047-0.0105 nm for the bond length of d1

Fig.2 Top and side views of the most stable adsorption of thiophene molecule on M(111)surfaces
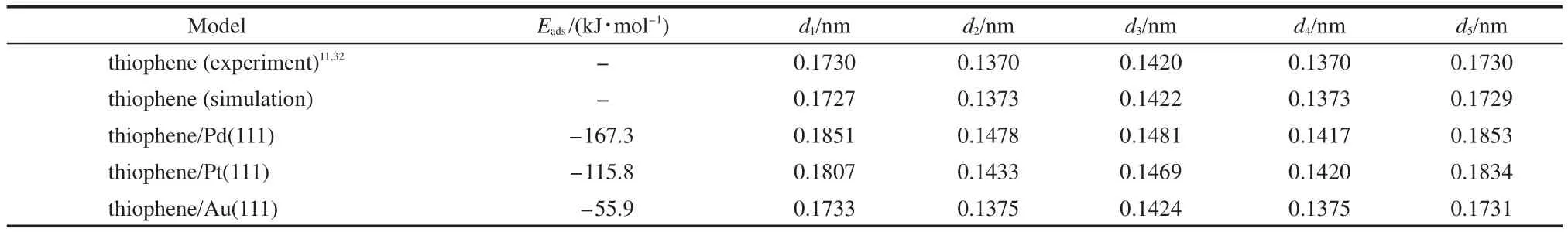
Table 2 Adsorption energies and structure parameters of thiophene molecule on M(111)surfaces for the most stable adsorption geometry
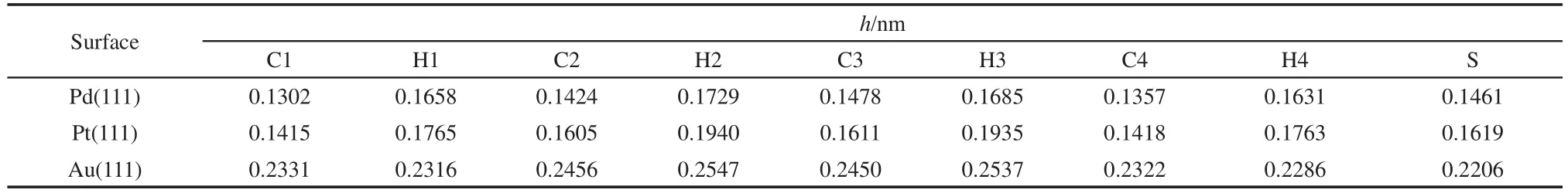
Table 3 Distance(h)from atoms of thiophene to M(111)surfaces for the most stable adsorption geometry
-d5,and the change of d5is the largest,which has been elongated from 0.1729 to 0.1834 nm.After the adsorption,structure relaxation of the top layer metal surface occurs and the S atom of thiophene transfers to the hollow site between the hcp and top sites.The H atoms of thiophene move upward and H2,H3 tilt up more obviously.Besides that,C1 and C4 atoms locate on the same plane and C2 and C3 atoms lie flush with the S atom.These results indicate that the ring plane of thiophene folds on the metal surface.Finally,the thiophene adsorbs on the Pt(111)surface by the atoms of C1,C4,and S.
When thiophene molecule adsorbs on Au-p-fcc-30°site,there is an elongation in the range of 0.0002-0.0006 nm for the bond length of d1
-d5.The d1value has been elongated from 0.1727 to 0.1733 nm,which increases obviously.After the adsorption,there is no obvious structure relaxation for the top layer metal surface and the S atom of thiophene shifts from the fcc site to the top site.All atoms of thiophene almost have no change and the ring plane tilts slightly.Finally,the thiophene adsorbs on theAu(111)surface with the atom of S.
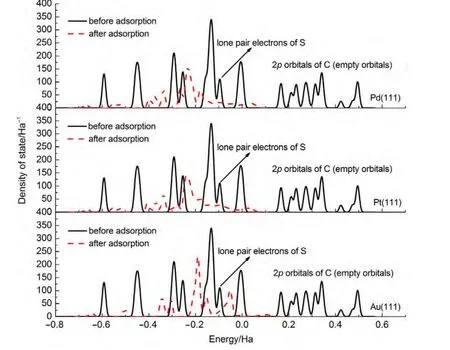
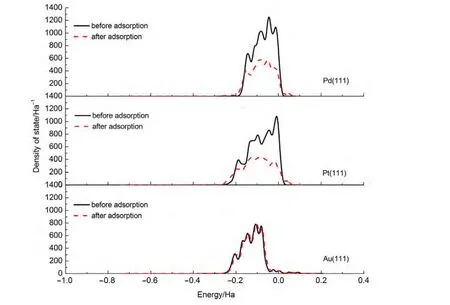
By comparing the most stable adsorption geometries of thiophene molecule on M(111)surface,we found that the order of thiophene molecule deformation is:Pd>Pt>Au.Meanwhile,the order of distance value between the thiophene and M(111)surface is:Pd Mullikenpopulation analysis33was done to reflect the charge distribution of the atoms and bonds in an adsorption system.In order to further analyze the adsorption of thiophene molecule on M(111)surface,Mulliken atomic charge populations of thiophene are summarized in the Table 4. Because the electronegativity of S atom is stronger than that of C atom,therefore the bonding electrons,especially the p electrons are biased toward the S atom in free thiophene,which increased the polarity of C―S bond.It can be seen from Table 4,atomic chargepopulationof Sis-0.143e,C1is-0.017e,C2is-0.028e,C3 is-0.029e,and C4 is-0.017e.The electron-withdrawing effectof S atom makes charge population of C1,C4 higher than those of C2,C3.The H atoms are positively charged,which would lead to the electric neutrality of total charge of thiophene. Table 4 Mulliken atomic charge populations of thiophene molecule at advantage adsorption site on the M(111)surface When thiophene adsorbs on Pd(111),Pt(111),or Au(111)surface,the atomic charge population of thiophene increases from 0.000e to 0.946e,0.682e,and 0.101e.It shows that the electrons have been transferred from the thiophene molecule to the M(111)surface,which indicates that the aromaticity of thiophene molecule has been damaged and the C atoms are characteristic of sp3hybridization.In addition,the increment of charge population implies that the interaction of thiophene and Pd(111)surface is the strongest,and the interaction of thiophene and Au(111)surface is the weakest,which is well consistent with the results of adsorption energies and the structure parameters. The charge density difference(CDD)34can be used to analyze the electron density redistribution after adsorption more directly.In order to give an intuitional insight into the electronic interactions between thiophene and M(111)surface,we also calculate the charge density difference of the most stable adsorption sits,as shown in Fig.3.It is defined as: where ρthiophene/M(111)is the charge density for the adsorbed systems;ρthiopheneand ρM(111)are the charge densities for the free thiophene and the non-interacting slab substrate,respectively.The red and the blue parts represent the accumulation and depletion electrons,respectively.It can be learned from Fig.3 that the charge density increases and a sort of nodal plane can be seen on metal surface when the adsorption occurs,which indicates that the interaction between the thiophene molecule and M(111)surface is more electrostatic than covalent.Besides that,by comparing the region of adsorption,we can also see that the order of bond strength for different metals are Pd,Pt then Au,this is the similar trend as shown in Mulliken population analysis. In order to highlight the electronic interactions between the thiophene molecule and M(111)surface,we calculated the PDOS for the p-orbitals of thiophene molecules and the d-orbitals of surface layer of M(111)surface before and after adsorption,as shown in Fig.4andFig.5.TheFermi level of the adsorption systems is set to 0 Ha. From PDOS of thiophene molecule before adsorption in Fig.4,it can be seen that the PDOS above Fermi level represents the lowest unoccupied molecular orbital(LUMO),while that between 0.0-0.8 Ha below the Fermi level represents the highest occupied molecular orbital(HOMO).The LUMO is contributed mainly by 2p orbitals of C atoms and the HOMO is collaboratively contributed by the p orbitals of S atom and C atoms.Furthermore,the peak at-0.5 Ha is attributed to the lone pair electrons of S atom.When thiophene molecule adsorbs on M(111)surface,the PDOS peaks shift to lower energy and the peak at the Fermi level disappears.These results proved that the lone pair electrons of S atom participate in forming π bond of thiophene and there is a strong interaction between π bond of thiophene and the d-orbitals of M(111)surface.In addition,the most visible change after thiophene adsorbed on M(111)surface is that the peaks on the bottom of LUMO decreased,or even disappeared.This means that,the electrons of M(111)surface transfer to the empty orbitals of thiophene molecule,its aromaticity has been destroyed,and the C atoms are characteristic of sp3hybridization,which is in good agreement with geometry analysis and charge population analysis. Fig.3 Charge density difference of thiophene molecule after adsorption on M(111)surfaces Fig.4 Partial density of state for the p-orbitals of thiophene molecules before and after adsorption at the most stable adsorption site on the M(111)surface Fig.5 Partial density of state for the d-orbitals of surface layer of M(111)surface before and after adsorption at the most stable adsorption site It can be seen from Fig.4 and Fig.5 that the PDOSs for the dorbitals of M(111)surface are intersect with Fermi level whether before or after adsorption,which reflects the metallicity of substrate.For the adsorption of thiophene on Au(111)surface,the PDOS for the d-orbitals almost does not change,which illustrates that the adsorption between thiophene and Au(111)surface is weak.For the adsorption of thiophene on Pd(111)and Pt(111)surfaces,the peaks of PDOS for the d-orbitals are sharp and high before adsorption.After the thiophene molecule adsorption on the Pd(111)or Pt(111)surface,the peaks of PDOS are reduced and obtuse,which reflects the stronger interaction between the π-bond of thiophene molecule and thed-orbitals of Pd(111)or Pt(111)surface.This interaction ultimately leads to the magnitude of the distortion in PDOS of thiophene molecule.After analyzing the PDOS and the charge population altogether,we can find that the donation and back-donation of electrons occur in the process of adsorption between thiophene molecule and the M(111)surface.This electron transfer process plays an important role in adsorption of thiophene on M(111)surface. In this paper,we performed a systematical study of the adsorption of thiophene on the M(111)(M=Pd,Pt,Au)surface by density functional theory coupled with periodic slab models. The parallel adsorption sites are more stable than the vertical adsorption sites.For Pd(111)and Pt(111)surfaces,the most stable adsorption structure is the parallel adsorption for the hollow site through the ring plane of thiophene,while,the most stable adsorption structure of Au(111)surface is the tilt adsorption for the top site through the S atom of thiophene.The order of adsorption energy is Pd(111)>Pt(111)>Au(111). After thiophene is adsorbed,all the bond length of which increased and the H atom is mostly like to attack the α-C of thiophene when the hydrodesulfurization reaction occurs.The structure of thiophene on Au(111)surface almost has no change,while for Pd(111)and Pt(111)surfaces,the H atom of thiophene tilts upward and its structure is distortional and folded.The aromaticity of thiophene is damaged and the C atoms are characteristic of sp3hybridization. The electrons of M(111)surface and thiophene are redistributed after adsorption.The electrons transfer from thiophene to M(111)surface and the order is Pd(111)>Pt(111)>Au(111).In addition,the electrons of M(111)surface are also back-denoted to the empty orbitals of thiophene molecule.This collaborative process plays an important role in adsorption of thiophene on M(111)surface. (1) Parola,V.L.;Testa,M.L.;Venezia,A.M.Appl.Catal.BEnviron.2012,119,248. (2) Li,J.;Huang,H.N.;Liang,W.H.;Gao,Q.;Duan,Z.Org.Lett.2013,2,282. (3) Urban,S.;Beiring,B.;Ortega,N.;Paul,D.;Glorius,F.J.Am.Chem.Soc.2012,134,15241.doi:10.1021/ja306622y (4) Zhang,B.Y.;Jiang,Z.X.;Li,J.;Zhang,Y.N.;Lin,F.;Liu,Y.;Li,C.J.Catal.2012,287,5.doi:10.1016/j.jcat.2011.11.003 (5) Rang,H.;Kann,J.;Oja,V.Oil Shale 2006,23,164. (6) Lu,W.T.;Chen,J.C.;Feng,J.;Yu,J.Rare Metal.Mat.Eng.2012,41,184. (7) Mittendorfer,F.;Hafner,J.J.Catal.2003,214,234.doi:10.1016/S0021-9517(02)00149-5 (8) Zaera,F.;Kollin,E.B.;Gland,J.L.Surf.Sci.1987,184,75.doi:10.1016/S0039-6028(87)80273-X (9)Zhu,H.Y.;Lu,X.Q.;Guo,W.Y.;Li,L.F.;Zhao,L.M.;Shan,H.H.J.Mol.Catal.A-Chem.2012,363-364,18. (10) Cocco,R.A.;Tatarchuk,B.J.Surf.Sci.1989,218,127.doi:10.1016/0039-6028(89)90623-7 (11) Terada,S.;Yokoyama,T.;Sakano,M.;Imanishi,A.;Kitajima,Y.;Kiguchi,M.;Okamoto,Y.;Ohta,T.Surf.Sci.1998,414,107.doi:10.1016/S0039-6028(98)00495-6 (12)Sato,H.;Ushiyama,S.;Sogo,M.;Aoki,M.;Shudo,K.;Sugawara,T.;Yanagisawa,S.;Morikawa,Y.;Masuda,S.Phys.Chem.Chem.Phys.2012,14,15412.doi:10.1039/c2cp42700a (13) Zhou,J.;Yang,Y.X.;Liu,P.;Camillone,N.;White,M.G.J.Phys.Chem.C 2010,114,13670.doi:10.1021/jp1025009 (14)Heermann,D.W.Computer Simulation Methods in Theoretical Physics;Springer-Verlag:Heidelberg,1990. (15) Leach,A.R.Molecular Modelling:Principles and Applications;Addison Wesley Longman Limitted Press:Essex,2001. (16) Sitamraju,S.;Janik,M.J.;Song,C.S.Top.Catal.2012,55,229.doi:10.1007/s11244-012-9807-1 (17) Callsen,M.;Atodiresei,N.;Caciuc,V.;Blugel,S.Phys.Rev.B 2012,86,1. (18)Wang,L.T.;Sun,Z.L.;Ding,Y.;Chen,Y.C.;Li,Q.;Xu,M.;Li,H.L.;Song,L.J.Appl.Surf.Sci.2011,257,7539.doi:10.1016/j.apsusc.2011.03.115 (19)Shi,W.;Zhang,L.Y.;Ni,Z.M.;Xiao,X.C.;Xia,S.J.RSC Adv.2014,4,27003. (20)Tang,F.W.;Guo,W.M.;Tang,N.N.;Pei,J.Y.;Xu,X.Acta Phys.-Chim.Sin.2013,29,2198.[唐法威,郭为民,唐楠楠,裴俊彦,许 旋.物理化学学报,2013,29,2198.]doi:10.3866/PKU.WHXB201307294 (21)Xiao,X.C.;Shi,W.;Ni,Z.M.Acta Phys.-Chim.Sin.2014,30,1456.[肖雪春,施 炜,倪哲明.物理化学学报,2014,30,1456.]doi:10.3866/PKU.WHXB201406091 (22)Ni,Z.M.;Shi,W.;Xia,M.Y.;Xue,J.L.Chem.J.Chin.Univ.2013,34,2353.[倪哲明,施 炜,夏明玉,薛继龙.高等学校化学学报,2013,34,2353.] (23) Ge,Q.;Jenkins,S.J.;King,D.A.Chem.Phys.Lett.2000,327,125.doi:10.1016/S0009-2614(00)00850-2 (24) Chen,Z.H.;Ding,K.N.;Xu,X.L.;Li,J.Q.Chin.J.Struct.Chem.2010,29,365. (25) Delley,B.J.Chem.Phys.2000,113,7756.doi:10.1063/1.1316015 (26) Perdew,J.P.;Chevary,J.A.;Vosko,S.H.;Jackson,K.A.;Pederson,M.R.;Singh,D.J.;Fiolhais,C.Phys.Rev.B 1992,46,6671.doi:10.1103/PhysRevB.46.6671 (27) White,J.A.;Bird,D.M.;Payne,M.C.;Stich,I.Phys.Rev.Lett.1994,73,1404.doi:10.1103/PhysRevLett.73.1404 (28) Monkhorst,H.J.;Pack,J.D.Phys.Rev.B 1976,13,5188.doi:10.1103/PhysRevB.13.5188 (29) Kittel,C.C.Solid State Physics;John Wiley&Sons:New York,1976. (30)Mai,S.W.;Zhou,G.D.;Li,W.J.Advanced Inorganic Structural Chemistry;Peking University Press:Beijing,2001.[麦松威,周公度,李伟基.高等无机结构化学.北京:北京大学出版社,2001.] (31)Atsushi,I.;Franck,D.;Jeayoung,L.;Kouhei,M.;Eika,Q.W.;Toshiaki,K.Appl.Catal.A-Gen.2005,289,163.doi:10.1016/j.apcata.2005.04.056 (32) Higai,S.;Nara,J.;Ohno,T.Surf.Sci.2006,600,685.doi:10.1016/j.susc.2005.11.033 (33) Mulliken,R.S.J.Chem.Phys.1955,23,1833.doi:10.1063/1.1740588 (34)Teng,B.T.;Zhao,Y.;Wu,F.M.;Wen,X.D.;Chen,Q.P.;Huang,W.X.Surf.Sci.2012,606,1227.doi:10.1016/j.susc.2012.04.0013.2 Charge population analysis

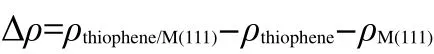
3.3 Density of states analysis

4 Conclusions
猜你喜欢
中国机械工程(2023年3期)2023-02-20 14:19:56
中国机械工程(2022年3期)2022-02-24 02:53:04
速读·上旬(2021年4期)2021-07-23 08:38:31
中国机械工程(2021年11期)2021-06-23 12:50:34
中国机械工程(2019年17期)2019-09-19 07:42:46
西部论丛(2018年11期)2018-10-19 09:11:24
校园英语·中旬(2016年12期)2017-01-19 18:24:29
校园英语·下旬(2016年2期)2016-03-18 10:23:20
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
