
云平台下基因-基因相互作用识别算法
2014-09-12 00:48:46刘桂霞李广力
吉林大学学报(理学版) 2014年3期
刘桂霞,李广力,李 涵
(吉林大学计算机科学与技术学院,长春130012)
云平台下基因-基因相互作用识别算法
刘桂霞,李广力,李 涵
(吉林大学计算机科学与技术学院,长春130012)
针对现有的快速方差分析算法进行并行可扩展性改进,设计一种高效的并行计算模型,并提出一种基于MapReduce模型的基因-基因相互作用识别算法——MR-ANOVA算法.该算法有效解决了现有基因-基因相互作用识别算法在海量数据规模下普遍存在计算复杂度过高的问题.实验结果表明,该算法充分利用了云平台的并行计算能力,随着数据量的增大,加速比逐渐接近于集群数量,可高效准确地完成基因-基因相互作用的识别.
基因-基因相互作用;MapReduce模型;云计算
1 基于方差分析的基因-基因相互作用识别算法
1.1 方差分析方法
方差分析方法[5]旨在将整体数据间的差异分为若干组内数据差异与组间数据差异,从而进行比较,判定某个小组是否为有效数据.由于多基因作用的影响,表现型在某种意义上非常复杂,而利用方差分析可有效分析连续的表现型,从而找出影响遗传变异的可能因素.因此方差分析被认为是一种度量单核苷酸多态与表现型间关联性的标准统计学方法,并常用于表现型关联性的定量研究中.但由于双位点相关性检测非常耗时,方差分析并不能很好地适用于全基因组范围内,因此要引入更高效的处理方法.快速方差分析算法,即FastANOVA算法[3],是一种结合大量置换检验,并运用批处理方式对单核苷酸多态对进行方差分析的优化算法.由于该算法利用相互独立的多组表现型值排列而得出方差分析的上界,筛选出了少数有意义的候选单核苷酸多态对,所以仅需在少数候选SNP对中进行方差分析即可,且不会丢失有意义的SNP对.此外,某些统计量对于每一组别是特定的,从而在不同的置换检验中仅需计算一次.
1.2 算法并行可拓展性的改进
FastANOVA算法在计算阈值Fα时使用了迭代方法,在计算中使用一个全局更新表不断对Fα进行更新,这种迭代更新的方法使算法的并行可扩展性较差.考虑到算法对Fα进行更新的操作只是一种优化操作,并不会对程序最终运行结果产生影响,所以在对算法的改进中本文使用局部的更新表替代全局更新表,使算法具有更好的并行可扩展性,并易于在云平台下实现并行化.
2 基于MapReduce模型的并行化算法
MapReduce模型处理数据的过程可抽象成两个函数:Map和Reduce.Map将问题分解成多个任务,Reduce将分解后多个任务的处理结果进行综合[6].程序开发者只需设计出该模型中的Map和Reduce函数,而并行编程中可能出现的各类复杂问题,如工作调度、容错处理和网络通信等,都会由MapReduce框架负责解决[7-8].
基于MapReduce的快速方差分析算法(即MR-ANOVA)分为两个阶段,每阶段将作为Hadoop云计算框架中的一个任务完成.假设Xi和Xj均代表基因型值,而Y代表表现型排列,{X1,X2,…,Xn}是n个被关注SNP的集合.第一阶段进行置换检验,Map函数将SNP分组,并尽可能平均的将分组发送至Reduce.Reduce函数接收分组,计算对于分组中给定的Xi在所有XiXj(i<j)中p种不同排列的F值.第一阶段结束后,通过对结果的排序查找操作可得出用于求解第二阶段的Fα值.第二阶段进行基因-基因相互作用的求解,Map函数将SNP分组,并尽可能平均的将分组发送至Reduce.Reduce函数接收分组,对于分组中给定的Xi,计算所有XiXj(i<j)的F值,并输出F>Fα的SNP对.
2.1 高效的并行计算模型
在云平台上进行并行计算时,理想情况是集群中每个节点处理同样多的数据,这样能使每个节点的运算时间基本相同,此时算法可得到最高效的并行计算效果.对于两位点的基因-基因相互作用识别,需要计算全部SNP对的F值[9].通常情况下,N个SNP两位点的基因-基因相互作用识别中,对于编号为i的SNP,需要计算XiXj(i<j≤n)的全部F值,共需要计算n-i个F值,随着i的不断增大,计算量逐渐减少,所以将每个Xi平均分配给集群中的节点进行计算无法取得良好的并行效果.针对该问题,本文提出一种两位点基因-基因相互作用识别的高效并行计算模型.
假设有m个节点用于计算,在该模型中,SNPi与SNPn-i+1将组成一个分组,每个分组的计算量为n,分组个数为n/2,每个节点将计算(n/2)/m个分组,此时每个节点的计算量会相对均衡,将有效提高算法在云平台下并行计算的效率,该并行模型如图1所示.本文将这种高效的并行计算模型应用到快速方差分析算法中,不仅解决了云平台中各节点负载均衡的问题,同时也显著提高了并行计算的效率.
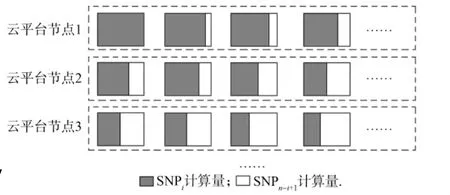
图1 并行计算模型Fig.1 Parallel computing model
2.2 MR-ANOVA算法
2.2.1 Map函数和Reduce函数 MR-ANOVA的第一阶段通过置换检验获取方差分析的阈值Fα,第二阶段根据阈值Fα求出所有F>Fα的SNP对.第一阶段Map函数的输入〈key,value〉键值对中,key为行号,value为Reduce的数量NR,Map函数将SNP按并行化模型进行分组,并尽可能均匀的将分组分配至各个Reduce中,将Reduce编号与SNP编号以〈key',value'〉键值对形式发送.第二阶段的Map函数与第一阶段的结构和功能相同.Map函数描述如下:

在MR-ANOVA第一阶段Reduce函数的输入〈key,value〉键值对中,key为Reduce编号,value为需要计算的SNP编号集合,Reduce函数的功能是对value中的每个编号i计算所有XiXj(i<j)全部排列(1~p)的F值,并发送〈key',value'〉键值对,其中key'为排列编号,value'为F值.第二阶段的Reduce函数中去掉了置换检验的步骤,其他步骤与第一阶段相同.Reduce函数描述如下:


2.2.2 算法的MapReduce框架 在MR-ANOVA算法中包含两个阶段,在Hadoop平台下,本文建立了job1和job2两个任务分别对应算法的两个阶段.在job1任务完成后,根据给定的Ⅰ型错误值和置换检验次数对job1任务的计算结果排序及查找操作,可获取第一阶段置换检验得出的Fα值.job2任务使用Fα值进行算法的第二阶段,找出符合条件的SNP对,并输出相关结果的信息.MR-ANOVA算法的整体框架如图2所示.

图2 MR-ANOVA算法框架Fig.2MR-ANOVA framework
3 实验结果及分析
本文使用Hadoop开源平台下的MapReduce框架,在本地集群上进行部署.本地集群由6台主机组成,其中1台主机作为master,5台主机作为slave,集群中每个节点的CPU频率均为2.80 GHz,内存容量为1 Gb.平台使用Hadoop1.0.4版本,操作系统为Ubuntu 10.04,使用JAVA语言实现.
在云平台下并行化算法的目的主要是为了解决基因-基因相互作用识别中密集计算的问题,本文以加速比(speedup)作为指标对算法性能进行检测.加速比是指同一算法在一台主机上的运行所需时间与并行化后在具有m个节点的集群上运行所需时间的比值,该指标常用于测试并行化算法运行效率提高的程度.加速比用Sp表示,表达式为Sp=Tv/Tm,其中Tm表示在具有m个节点的集群上完成MRANOVA算法所花费的时间;Tv表示在单个主机上运行算法所花费的时间.
本文实验使用的测试数据为来自Broad/MIT(http://www.broadinstitute.org/)的小鼠基因数据,利用NPUTE填补缺失的数据[10],并通过 Jackson Lab(http://jaxmice.jax.org/)查询了小鼠对应的Cardiovascular(blood pressure)表现型.为了更好体现MR-ANOVA算法的优势,实验使用不同规模的集群运行算法.实验中,不同的Ⅰ型错误值α也将导致出现不同的结果.实验使用的程序参数如下:Ⅰ型错误值α=0.1,个体数为35个,置换检验次数为100次.测试的SNP数量分别为1 000,2 000,5 000和10 000.

图3 测试结果Fig.3 Experimental results
MR-ANOVA算法利用 Hadoop完成 Map与Reduce的任务,最终完成基因-基因相互作用的识别过程,算法加速比测试结果如图3所示.由图3可见,随着SNP数量的不断增大,Hadoop平台可有效减小算法的时间消耗,加速比有显著提高. Hadoop平台节点间的数据传输及任务分配有一定的开销,当SNP数量较小时,集群运算所产生的加速效果并不明显.加速比随着集群规模增大而不断增大,可见随着节点数量的增大,算法时间消耗将进一步减少.
实验结果表明,本文提出的MR-ANOVA算法更适用于大规模数据计算,即当需要分析的SNP数量越大时,并行化加速效果越明显.其主要原因在于数据量较大时,Hadoop平台下数据操作的开销所占总时间的比例较小.MR-ANOVA算法利用云平台减轻了基因-基因相互作用识别算法密集计算的负担,是一种高效的基因-基因相互作用识别算法.
综上所述,本文提出的MR-ANOVA算法是对FastANOVA算法的一种并行化改进,有效减少了基因-基因相互作用识别算法中密集计算的时间消耗.MR-ANOVA算法充分利用了云平台的并行计算能力,可更高效完成基因-基因相互作用的识别.
[1] ZHANG Xiang,ZOU Fei,WANG Wei.FastChi:An Efficient Algorithm for Analyzing Gene-Gene Interactions[J]. Pacific Symposium on Biocomputing,2009,14:528-539.
[2] ZHANG Xiang,HUANG Shunping,ZOU Fei,et al.Tools for Efficient Epistasis Detection in Genome-Wide Association Study[J].Source Code for Biology and Medicine,2011,6(1):1.
[3] ZHANG Xiang,ZOU Fei,WANG Wei.Fastanova:An Efficient Algorithm for Genome-Wide Association Study[C]// Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining.New York: ACM,2008:821-829.
[4] 李建江,崔健,王聃,等.MapReduce并行编程模型研究综述[J].电子学报,2011,39(11):2635-2642. (LI Jianjiang,CUI Jian,WANG Dan,et al.Survey of MapReduce Parallel Programming Model[J].Acta Electronica Sinica,2011,39(11):2635-2642.)
[5] Bolton S.Analysis of Variance[J].Pharmaceutical Statistics:Practical and Clinical Applications,1997,737: 265-325.
[6] 李成华,张新访,金海,等.MapReduce:新型的分布式并行计算编程模型[J].计算机工程与科学,2011,33(3):129-135.(LI Chenghua,ZHANG Xinfang,JIN Hai,et al.MapReduce:A New Programming Model for Distributed Parallel Computing[J].Computer Engineering&Science,2011,33(3):129-135.)
[7] 刘鹏.云计算[M].2版.北京:电子工业出版社,2011:21-24.(LIU Peng.Cloud Computing[M].2nd ed. Beijing:Publishing House of Electronics Industry,2011:21-24.)
[8] 刘鹏.实战Hadoop:开启通向云计算的捷径[M].北京:电子工业出版社,2011:60-62.(LIU Peng.Action in Hadoop:Open the Shortcut to Cloud Computing[M].Beijing:Publishing House of Electronics Industry,2011: 60-62.)
[9] Wang Z,Wang Y,Tan K L,et al.eCEO:An Efficient Cloud Epistasis Computing Model in Genome-Wide Association Study[J].Bioinformatics,2011,27(8):1045-1051.
[10] Roberts A,McMillan L,Wang W,et al.Inferring Missing Genotypes in Large SNP Panels Using Fast Nearest-Neighbor Searches over Sliding Windows[J].Bioinformatics,2007,23(13):i401-i407.
(责任编辑:韩 啸)
Algorithms for Detecting Gene-Gene Interactions Based on Cloud Platform
LIU Guixia,LI Guangli,LI Han
(College of Computer Science and Technology,Jilin University,Changchun 130012,China)
The authors proposed an optimized algorithm for detecting gene-gene interactions based on MapReduce model,namely,MR-ANOVA.Compared with the traditional FastANOVA algorithm,this algorithm puts forward the concept of parallel processing during which an efficient parallel computing model is used.This improvement can make the problem of high computational complexities with the large-scale data of the existing algorithms solved.Analyzing results of the experiment,we can draw the following conclusion: MR-ANOVA algorithm can make the best use of the promising power of parallelism computation of the cloud platform.As the scale of the data becomes larger,the speedup is more close to the number of clusters.Thus,this optimized algorithm can detect epistatic interaction more efficiently.
gene-gene interaction;MapReduce model;cloud computing
TP311.1
A
1671-5489(2014)03-0546-05
10.13413/j.cnki.jdxblxb.2014.03.26
全基因组关联研究(genome-wide association studies,GWAS)是在全基因组范围内对单核苷酸多态(single nucleotide polymorphism,SNP)与表现型间潜在关系的研究,它能有效检测哪些遗传因素可导致个体间性状形成的差异,对于人类疾病等生物研究领域有重要意义.传统的全基因组关联研究仅关注于单个单核苷酸多态与表现型间的关系,而未考虑SNP间相互作用并共同改变表现型的情况,这种不同基因位点同时控制相同性状的现象被称为基因-基因相互作用.目前,已有很多应用于基因-基因相互作用的识别算法,如FastChi算法[1]、利用凸优化的COE算法[2]和基于方差分析的FastANOVA算法[3]等.目前的大部分算法虽减小了搜索空间,但仍不能真正解决大量的单核苷酸多态对相互作用产生的密集计算问题.MapReduce是一种云平台下的并行编程模型,能连接大量主机的计算资源,从而形成规模巨大的集群,适用于处理海量数据的并行计算[4].本文使用Hadoop平台,利用MapReduce模型对FastANOVA算法进行并行化改进.
2014-03-10.
刘桂霞(1963—),女,汉族,博士,教授,博士生导师,从事计算智能理论、云计算技术及其应用的研究,E-mail:liugx @jlu.edu.cn.通信作者:李广力(1992—),男,汉族,从事计算智能理论、云计算技术及其应用的研究,E-mail:calculatinggod@ foxmail.com.
国家自然科学基金(批准号:61373051;61175023)、吉林省科技发展计划重点项目(批准号:20140204004GX)和吉林大学“大学生创新创业训练计划”项目(批准号:2013B53205).
猜你喜欢
系统工程学报(2021年4期)2021-12-21 06:21:08
新世纪智能(高一语文)(2020年12期)2020-06-01 08:14:20
军事运筹与系统工程(2019年4期)2019-09-11 06:39:58
新课程·下旬(2018年9期)2018-11-14 12:49:12
电子制作(2018年11期)2018-08-04 03:25:40
中国交通信息化(2017年3期)2017-06-08 06:09:28
知识就是力量(2017年2期)2017-01-21 18:29:36
中学生理科应试(2016年7期)2016-05-14 15:39:39
中学生理科应试(2015年12期)2016-01-11 10:34:20
高中生学习·高三版(2015年2期)2016-01-04 22:41:24
