
HPLC法测定胃健宁胶囊中和厚朴酚及厚朴酚的含量
2014-09-05 06:33:24叶锦雄刘柳毅
药学研究 2014年4期
叶锦雄,刘柳毅
(1.广州市东升医院,广东 广州 510140;2.广州中医药大学中药学院,广东 广州 510006)
胃健宁胶囊由厚朴、木香、砂仁、佛手、香附(醋制)、大腹皮等药组成。厚朴是处方中的君药,故对厚朴中的和厚朴酚与厚朴酚进行含量测定,现将结果报道如下:
1 仪器与试药
1.1 仪器 日本岛津:SPD-10A检测器、LC-10AT泵、N2000色谱工作站,AUW120D电子天平。
1.2 试剂 乙腈(分析纯,批号:20121204,江苏强盛化工有限公司);甲醇(分析纯,批号:20120102,天津市百世化工有限公司)。厚朴酚对照品(批号:110729-201210,中国药品生物制品检定所);和厚朴酚对照品(批号:110756-201210,中国药品生物制品检定所)。
2 方法与结果
2.1 对照品溶液的制备 精密称取和厚朴酚对照品10 mg,置50 mL量瓶中,加甲醇使溶解并稀释至刻度,摇匀,作为A液;另精密称取厚朴酚对照品10 mg,置50 mL量瓶中,加甲醇使溶解并稀释至刻度,摇匀,作为B液。分别精密吸取A液和B液各1 mL,置10 mL量瓶中,加甲醇使溶解并稀释至刻度,摇匀,即得。
2.2 供试品溶液的制备 取胃健宁胶囊20粒,除去囊壳,研细,取粉末约4 g,精密称定,置具塞锥形瓶中,精密加入氯仿50 mL,密塞,称定重量,超声处理1.5 h,称定重量,用氯仿补足减失的重量,摇匀,滤过,精密量取续滤液25 mL,蒸干,残渣用甲醇溶解并转移到5 mL容量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
2.3 缺厚朴阴性溶液制备 取不含厚朴的全部处方药材,按胃健宁胶囊工艺及上述供试品溶液的方法制备阴性对照液。
3 测定法
3.1 色谱条件 色谱柱:Kromasil C18柱(4.6 mm×250 mm,5 μm),流动相:乙腈 -水 -冰醋酸(60∶40∶1)(V∶V∶V),流速:1.000 mL·min-1,检测波长:294 nm,进样量:10 μL。
3.2 色谱条件与系统适用性试验 分别取对照品溶液、供试品溶液、阴性对照溶液各10 μL,注入色谱仪,测定,记录色谱图,结果见图1~3。由图可见,供试品色谱中,在与对照品色谱相应位置上,有相同保留时间的色谱峰,阴性试验无干扰。保留时间:厚朴酚峰约17 min,和厚朴酚峰约7 min,分离度大于1.5,阴性对照溶液无干扰。
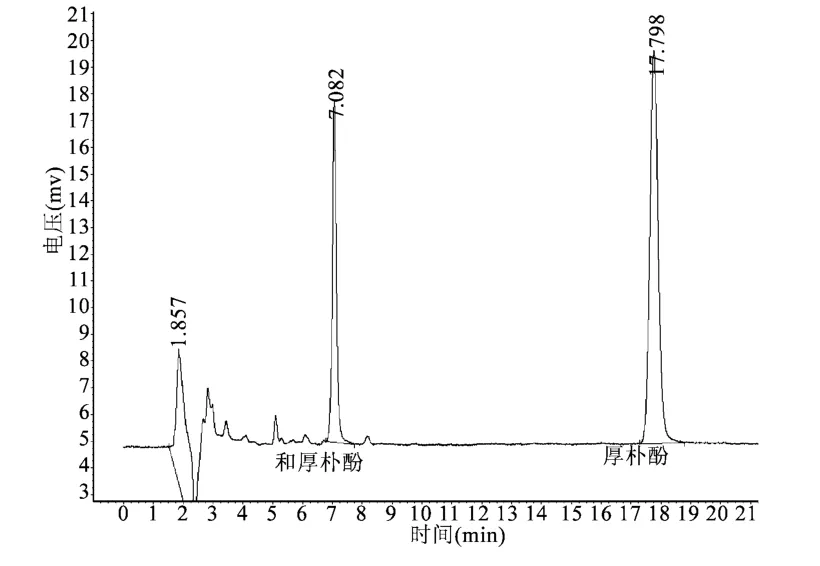
图1 对照品色谱图
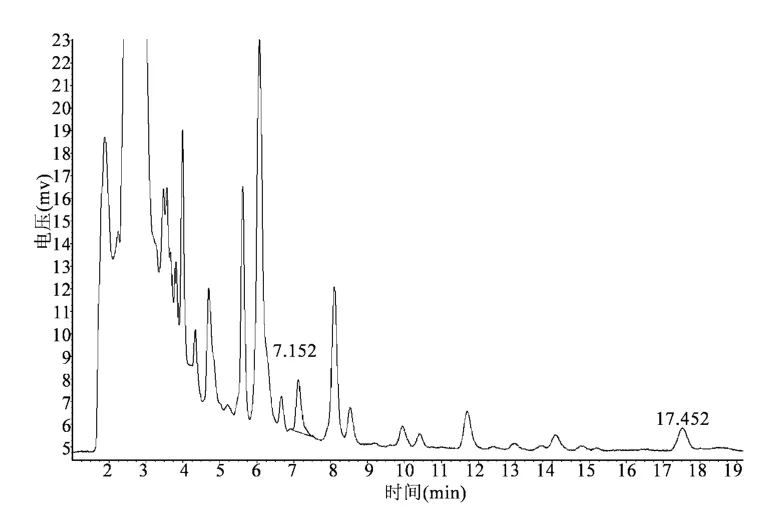
图2 供试品色谱图
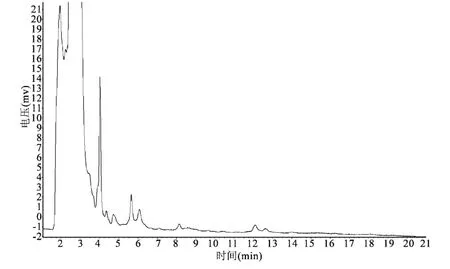
图3 阴性对照品色谱图
3.3 线性实验 精密吸取厚朴酚及和厚朴酚对照品液各 1、2、4、8、16 μL,注入色谱仪,记录色谱图,以峰面积积分值(Y)对对照品量(X)进行处理,和厚朴酚回归方程Y=6.45×105X-4.56×103,r=0.9996(n=5);厚朴酚回归方程Y=1.40×106X-1.67×103,r=0.9998(n=5)。结果表明,和厚朴酚在0.02~0.32 μg范围内线性关系良好;厚朴酚在0.0196~0.3136 μg范围内线性关系良好。
3.4 精密度试验 精密进样同一对照品溶液20 μL,连续测定5次,厚朴酚的平均峰面积为62997.93,RSD为1.61%,和厚朴酚的平均峰面积为61100.9024,RSD为1.10%,结果表明该方法的精密度良好。
3.5 稳定性试验 取样品适量,照含量测定项下方法测定,于配制后0、2、4、8、12 h,分别取样测定其含量。和厚朴酚的RSD为0.88% ,厚朴酚的RSD为1.96%,结果表明被测溶液的稳定性良好。
3.6 重复性试验 取样品6份,按含量测定项下方法分别测定,6份样品实验和厚朴酚的峰面积平均的RSD为2.6%,厚朴酚的峰面积平均值的RSD为2.1%。结果表明重复性良好。
3.7 加样回收率试验 取重复性试验的同一批号(120406)样品20粒,除去外壳,研细,取粉末约2 g,精密称定,置具塞锥形瓶中,分别精密加入一定量的和厚朴酚对照品及厚朴酚对照品,按供试品溶液的制备方法共制备3份,每份测定3次,计算回收率。和厚朴酚的平均回收率为96.58%,RSD为1.78%,厚朴酚的平均回收率为97.29%,RSD为1.39%,结果表明,含量回收率良好,见表1。
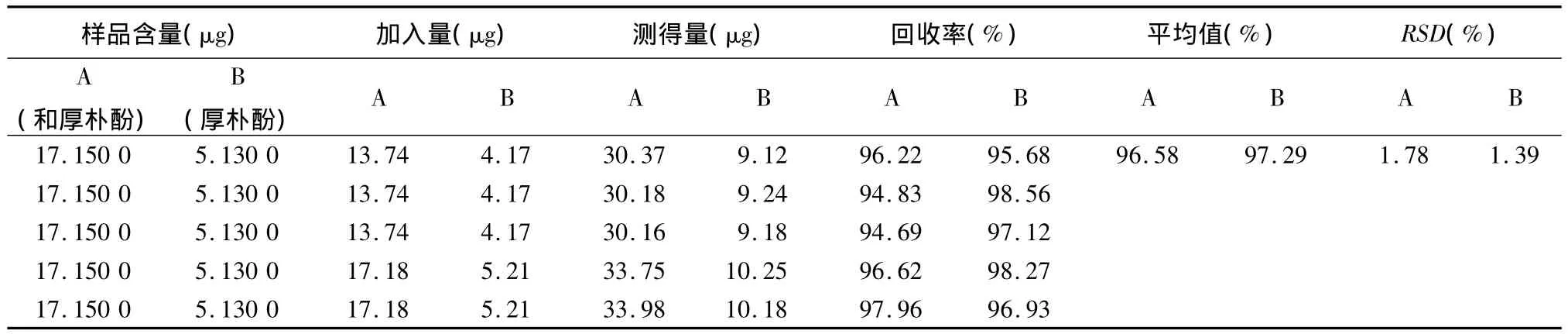
表1 胃健宁胶囊中和厚朴酚及厚朴酚的加样回收率试验结果(n=9)
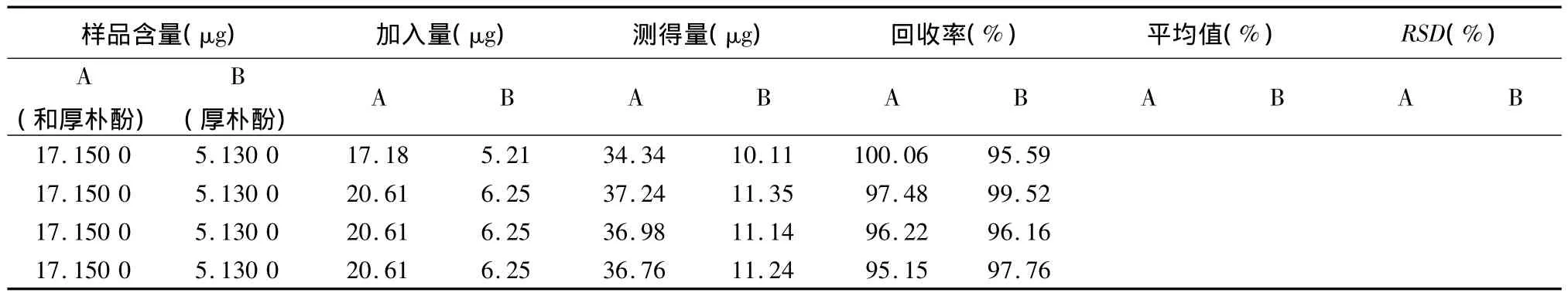
续表1:
3.8 样品含量测定 取3批样品(批号:120406、120408、120410),照“2.2”项下的方法制备供试品溶液,分别精密吸取对照品溶液与供试品溶液各10 μL,注入液相色谱仪测定,结果见表2。
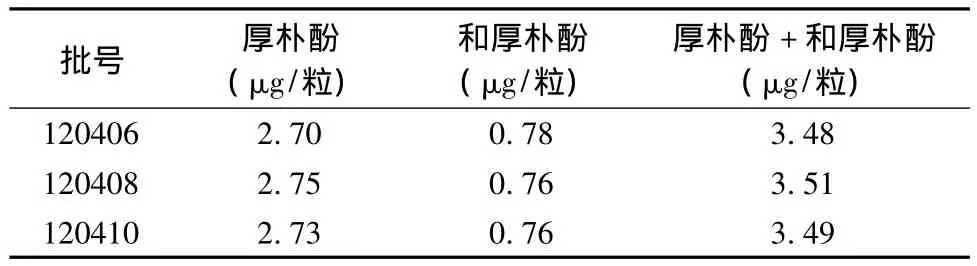
表2 样品含量测定结果(n=3)
由3批样品含量测定的结果可知,和厚朴酚及厚朴酚的重量平均值为每粒3.49 μg,考虑到生产中药材质量、制剂生产、贮藏等因素影响,暂定其制剂中和厚朴酚及厚朴酚含量不得低于每粒3 μg。
4 讨论
参照《中国药典》2010年版(一部)厚朴的含量测定方法[1]选择流动相甲醇 - 水(78∶22)进样,结果阴性对照有干扰。参考相关文献[2~6]采用流动相为乙腈-水-冰醋酸(60∶40∶1)进样,试验结果表明,在此条件下和厚朴酚、厚朴酚的保留时间适宜,与相邻的色谱峰可保持良好的分离度,适合对和厚朴酚、厚朴酚的含量测定。
制备供试品溶液采用甲醇提取样品时干扰较多,最后确定用三氯甲烷提取蒸干,残渣用甲醇溶解,分离效果较好。该方法结果准确可靠,重现性好,可用于胃健宁胶囊中和厚朴酚及厚朴酚的含量测定。本研究的不足之处就是待测成分含量较低,所得结果相对误差偏大。由于处方组成药材较多,各种成分含量均较低,且干扰成分多,但该方法对本复方中药制剂的质量控制仍具有重要意义。
[1]国家药典委员会.中华人民共和国药典2010年版(一部)[S].北京:中国医药科技出版社,2010:235.
[2]孟蕾蕾,李梦,王国平.HPLC法测定藿香正气胶囊中厚朴酚与和厚朴酚的含量[J].齐鲁药事,2007,26(8):466-467.
[3]魏刚.高效液相色谱法测定藿香正气丸中厚朴酚与和厚朴酚的含量[J].湖北中医杂志,2011,33(2):70-71.
[4]魏蓉,项菲.高效液相色谱法测定舒肝快胃丸中厚朴酚与和厚朴酚的含量[J].中南药学,2011,9(11):847.850.
[5]高辉,王雪萍.厚朴温中丸中厚朴酚与和厚朴酚含量测定的研究[J].中华中医药学刊,2008,26(2):425-426.
[6]赵彩霞,闫辉.麻仁丸中厚朴酚与和厚朴酚测定方法的研究[J].湖南中医药大学学报,2012,32(6):15-17.
猜你喜欢
煤炭与化工(2024年2期)2024-03-30 08:09:52
化工管理(2022年14期)2022-12-02 11:46:54
中国土壤与肥料(2021年5期)2021-12-02 01:04:34
今日农业(2020年22期)2020-12-14 16:45:58
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
中成药(2018年12期)2018-12-29 12:26:02
中国化肥信息(2016年27期)2016-05-17 04:25:18
Asian Journal of Urology(2015年3期)2015-12-16 14:42:15
电力需求侧管理(2014年4期)2014-03-20 13:35:51
化工生产与技术(2014年6期)2014-02-27 13:42:11
