
八氢吡咯并[3,4-c]吡咯-2-碳酸叔丁酯的合成*
2014-08-30 02:26:33朱思兰张伟萍史海健
合成化学 2014年5期
朱思兰,朱 晶,张伟萍,陈 程,史海健
(南京工业大学 化学化工学院,江苏 南京 210009)
·研究简报·
八氢吡咯并[3,4-c]吡咯-2-碳酸叔丁酯的合成*
朱思兰,朱 晶,张伟萍,陈 程,史海健
(南京工业大学 化学化工学院,江苏 南京 210009)
建立了一种构建八氢吡咯并[3,4-c]吡咯烷类化合物母体双环结构的快速有效方法。以苄胺为原料,与氯甲基三甲基硅烷反应制得仲胺化合物(2);2与甲醇和甲醛反应得亚甲胺叶立德前体化合物(3);在三氟乙酸的诱导作用下,3与马来酰亚胺经1,3-偶极环加成反应得顺式加成产物(4);4经氢化铝锂还原得5-苄基八氢吡咯并[3,4-c]吡咯(5);5经Boc保护氨基后用氢氧化钯碳催化加氢脱苄合成了八氢吡咯并[3,4-c]吡咯-2-碳酸叔丁酯,总收率35%,其结构经1H NMR,IR和MS确证。
八氢吡咯并[3,4-c]吡咯;亚甲胺叶立德;环加成;合成
八氢吡咯并[3,4-c]吡咯烷类化合物(Ⅰ,Chart 1)是良好的α4β2和α7nAChR子类型激动剂。只需简单改变母体双环结构Ⅰ的取代基R1和R2,就会对不同的受体子类型产生高选择性[1-3]。同时,也有文献报道一些基于Ⅰ的衍生物根据其功能不同被用于各种医学试验中,比如作为偏头痛药、抗菌素、羟色胺再摄取抑制剂、凝血酶抑制剂、抗焦虑药等[4]。由于Ⅰ的双环结构具有刚性特征,且易于在N上引入各种官能团以满足不同的研究需要,因此其在药物设计中是非常有用的母体骨架,广泛存在于天然生物碱中,具有重要的研究价值。
利用亚甲胺叶立德(C=N+-C-)与亲偶极体的1,3-偶极环加成是构建吡咯环结构的直接快速方法。构建该双环骨架的主要方法有Padwa研究组[5-6]利用氟化银、氟化锂为诱导试剂及Fray研究组[7]以TFA为诱导试剂等。
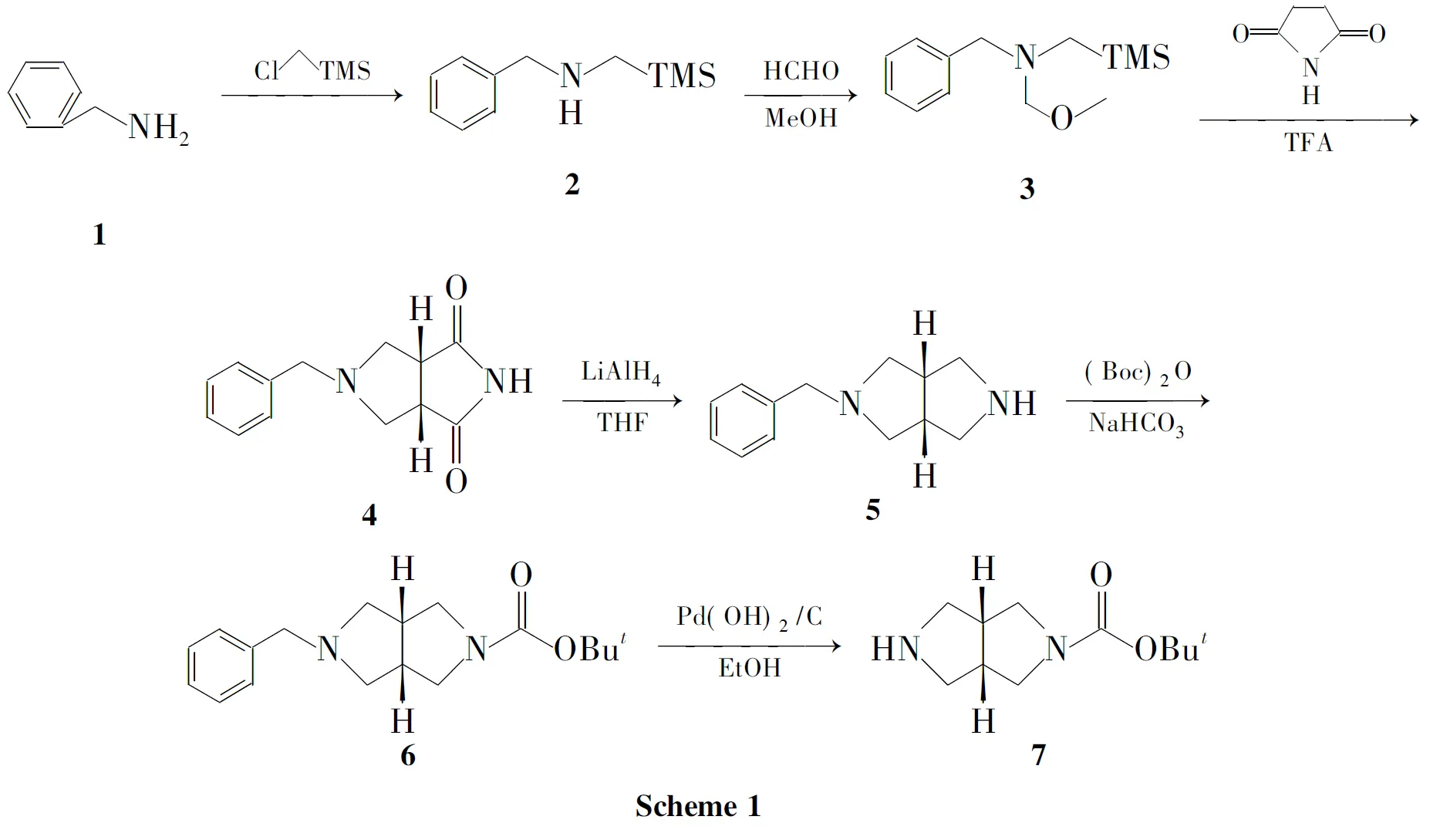
本文设计了一种合成八氢吡咯并[3,4-c]吡咯-2-甲酸叔丁酯的快速有效方法。以苄胺(1)为原料,与氯甲基三甲基硅烷反应制得仲胺化合物N-[(三甲基硅基)甲基]苄胺(2);2与甲醇和甲醛反应得亚甲胺叶立德前体化合物——N-苄基-N-(甲氧基甲基)-N-[(三甲基硅基)甲基]胺(3);在TFA的诱导作用下,3与马来酰亚胺经1,3-偶极环加成反应制得顺式加成产物5-苄基八氢吡咯并[3,4-c]吡咯-1,3-二酮(4);4经氢化铝锂还原得5-苄基八氢吡咯并[3,4-c]吡咯(5);5经Boc保护氨基后用氢氧化钯碳催化加氢脱苄合成了八氢吡咯并[3,4-c]吡咯-2-碳酸叔丁酯(7,Scheme 1),总收率35%,其结构经1H NMR,IR和MS确证。
1 实验部分
1.1 仪器与试剂
X-4型数字显微熔点仪(温度未校正);ACF-300Bruker型和ACF-400Bruker型核磁共振仪(CDCl3为溶剂,TMS为内标);Avatar-360型傅立叶红外光谱仪(KBr压片);Agilent1100型ZAB-HS质谱仪。
所用试剂均为分析纯或化学纯。
1.2 合成
(1)2的合成
在反应瓶中加入氯甲基三甲基硅烷12mL(86.08mmol)和118.81mL(172.17mmol),搅拌下于160℃反应5h(产生大量白色固体,TLC监测)。冷却至室温,过滤,滤饼用正己烷(3×50mL)洗涤,合并滤液和洗液,用水(2×50mL)洗涤,无水Na2SO4干燥,减压蒸除溶剂得淡黄色油状液体213.24g,收率79%;1H NMRδ:0.05(s,9H),2.08(s,2H),3.83(s,3H),7.23~7.35(m,5H)。
(2)3的合成
于0℃,在反应瓶中加入37%甲醛6.45mL(87.50mmol)和甲醇3.54mL(87.50mmol),搅拌使其呈均匀;缓慢滴加213.96g(72.20mmol),滴毕,于0℃~5℃反应1h;于10℃~15℃反应3h(TLC监测)。加入K2CO38g,搅拌2h,倾出有机层,加入K2CO31g,搅拌15min,有机层倾出,残余固体K2CO3用乙醚洗涤,合并有机层和洗液,用无水Na2SO4干燥,减压蒸除溶剂得淡黄色油状液体粗品,经减压蒸馏纯化得无色油状液体314.63g,收率85%;1H NMRδ:0.09(s,9H),2.23(s,2H),3.29(s,3H),3.80(s,2H),4.04(s,2H),7.26~7.36(m,5H)。
(3)4的合成
氮气保护,于0℃在反应瓶中依次加入马来酰亚胺0.98g(10.10mmol)和二氯甲烷23mL,搅拌使其溶解;加入TFA 0.08mL(1.08mmol),缓慢滴加33.12g(13.14mmol)的二氯甲烷(15mL)溶液,滴毕,自然升至室温,反应16h(TLC监测)。减压蒸除溶剂,残余物用CH2Cl235mL溶解,用饱和NaHCO3溶液(2×10mL)洗涤,水相用CH2Cl2(2×15mL)萃取,合并有机相,依次用饱和食盐水(15mL)洗涤,无水Na2SO4干燥,减压蒸除溶剂得淡黄色蜡状物42.25g,收率97%;1H NMRδ:2.32~2.43(m,2H),3.18(d,J=9.8Hz,2H),3.20~3.26(m,2H),3.59(s,2H),7.13~7.35(m,5H);IRν: 3435,1701,839,741,697cm-1。
(4)5的合成
氮气保护,于0℃在反应瓶中依次加入LiAlH41.11g(29.31mmol)和THF 30mL,搅拌使其呈混悬液;缓慢滴加42.25g(9.77mmol)的THF(20mL)溶液,滴毕,于室温反应30min;回流反应4h(TLC监测)。冷却至0℃,依次缓慢滴加水2.5mL,15%NaOH溶液2.5mL和水7.5mL淬灭反应,过滤,滤饼用乙酸乙酯(3×50mL)洗涤,合并滤液和洗液,减压蒸除溶剂得淡黄色蜡状物51.78g,收率90%;MSm/z(%): 203{[M+H]+,100}。
(5)5-苄基八氢吡咯并[3,4-c]吡咯-2-碳酸叔丁酯(6)的合成
在反应瓶中依次加入THF 20mL和51.78g(8.80mmol),搅拌使其溶解;加入(Boc)2O 2.34g(10.70mmol)和饱和NaHCO3溶液4.3mL,于室温反应过夜(有气体放出,不密闭,TLC监测)。加水(7mL)淬灭反应,加入乙酸乙酯20mL,分液,水相用乙酸乙酯(2×15mL)萃取,合并有机相,用无水Na2SO4干燥,减压蒸除溶剂得黄色油状液体,经硅胶快速柱层析[梯度洗脱剂:V(石油醚)∶V(乙酸乙酯)=10∶1~1∶1]纯化得淡黄色油状液体61.63g,收率61%;1H NMRδ:1.39(s,9H),2.25~2.36(m,2H),2.56~2.59(m,2H),2.64~2.76(m,2H),3.07~3.25(m,2H),3.37~3.48(m,2H),3.50(s,2H),7.15~7.23(m,5H);MSm/z(%): 303{[M+H]+,100}。
(6)7的合成
在10mL单口瓶中依次加入6140mg(0.46mmol),10%Pd(OH)2/C 140mg和乙醇7mL,充氮气2遍,通氢气,搅拌下于室温(常压)反应过夜。硅藻土助滤,滤饼用甲醇(2×15mL)洗涤,合并滤液和洗液,减压蒸除溶剂得无色油状液体795mg,收率97%;1H NMRδ:1.46(s,9H),2.65~2.68(dd,J=15.2Hz,3.6Hz,2H),2.83~2.84(m,2H),3.08~3.10(m,2H),3.20~3.23(dd,J=14.8Hz,3.4Hz,2H),3.52~3.55(m,2H);MSm/z(%): 213{[M+H]+,100}。
2 结果与讨论
2.1 合成与表征
1与氯甲基三甲基硅烷反应得2,反应机理为氯甲基三甲基硅烷中与氯相连的碳具有亲电性,进攻底物苄胺中带有孤对电子的氮,脱除一分子HCl后形成2。
2用甲醇和甲醛处理得3,反应机理为:甲醛分子中的羰基碳进攻2中带有孤对电子的氮,脱除氢氧根负离子后形成亚胺中间体,接着甲醇分子中带有孤对电子的氧进攻亚胺中间体中缺电子碳中心,脱除氢离子后生成3。
3与马来酰亚胺经1,3-偶极环加成反应构建双环骨架,经五元环过渡态,顺式加成立体选择得顺式4。4经LiAlH4还原得5,收率90%。
5的氨基保护反应,使用(Boc)2O引入Boc保护氨基,定量获得6。
用氢氧化钯碳催化加氢对6进行脱苄反应,于室温、常压下合成7,收率97%。
7的1H NMR分析表明,与6相比,低场苯环上5个氢及与苯环相连的亚甲基上两个氢质子信号消失,说明成功脱除苄基;δ2.66处的dd峰为7-位和8-位的两个氢质子,高场δ1.46处的单峰归属叔丁氧羰基中三个甲基上的9个氢质子;其余氢的个数和吸收峰位置均与7结构匹配,且与文献值[8]相吻合。
3 结论
以苄胺为起始原料原料,经两步反应合成亚甲胺叶立德前体化合物(3);在TFA的诱导作用下,3与马来酰亚胺经1,3-偶极环加成一步构建双环骨架,得顺式加成产物;经LiAlH4还原酰亚胺到胺,再用二碳酸二叔丁酯对氨基进行Boc保护,最后脱除苄基即得目标产物八氢吡咯并[3,4-c]吡咯-2-碳酸叔丁酯,采用氢氧化钯碳催化加氢脱苄的方法,总收率35%。
该合成路线新颖实用、简便易行,是构建八氢吡咯并[3,4-c]吡咯烷类化合物母体双环结构的快速有效方法,为相关药物的研发打下了基础。
[1] Arneric S P,Holladay M,Williams M.Neuronal Nicotinic Receptors:A perspective on two decades of drug discovery research[J].Biochem Pharmacol,2007,(74):1092-1101.
[2] D′Hoedt D,Bertrand D.Nicotinic acetylcholine receptors:An overview on drug discovery[J].Expert Opin Ther Targets,2009,(13):395-411.
[3] Olincy A,Harris J G,Johnson L L,etal.Proof-of-concept trial of an alpha7nicotinic agonist in schizophrenia[J].Arch Gen Psychiatry,2006,(63):630-638.
[4] Lee E K,Melville C R,Rotstein D.Heterocyclic antiviral compounds[P].US WO 2005121145,2005.
[5] Padwa A,Chen Y Y,Dent W,etal.Synthetic application of cyanoaminosilanes as azomethine ylide equivalents[J].J Org Chem,1985,(50):4006-4014.
[6] Padwa A,Dent W.N-bwenzyl-N-methoxymethyl-N-(trimethylsilyl)methylamine as an azomethine ylide equivalent:2,6-Dioxo-1-phenyl-4-benzyl-1,4-diazabicyclo[3.3.0]octane[J].Org Syntheses,1993,(8):231.
[7] Fray A H,Meyers A I.Diastereoselection in azomethine ylide cycloaddition reactions with unsaturated chiral bicyclic lactams[J].J Org Chem,1996,61:3362-3374.
[8] Bunnelle W H,Tietje K R,Frost J M,etal.Octahydropyrrolo[3,4-c]pyrrole:A diamine scaffold for construction of eitherα4β2orα7-selective nicotinic acetylcholine receptor(nAChR)ligands.Substitutions that switch subtype selectivity[J].J Med Chem,2009,(52):4126-4141.
SynthesisofOctahydropyrrolo-[3,4-c]pyrrole-2-carboxylicAcidTert-butylEster
ZHU Si-lan, ZHU Jing, ZHANG Wei-ping, CHEN Cheng, SHI Hai-jian
(College of Chemistry and Chemical Engineering,Nanjing Tech University,Nanjing 210009,China)
A simple and effective method to construct the bicyclic structure of octahydropyrrolo[3,4-c]pyrroles was developed.The secondary amine(2)was prepared by the reaction of benzylamine with chloromethyltrimethylsilane.The azomethine ylide precursor compound(3)was synthesized by the reaction of2with formaldehyde and methanol.In the presence of trifluoroacetic acid,the bicyclic skeleton was built by 1,3-dipolar cycloaddition reaction of3with maleimide to afford 5-benzyloctahydropyrrolo[3,4-c]pyrrole-1,3-dione(4).4was reduced by LiAlH4and then the amino group was protected by Boc.At last,octahydropyrrolo-[3,4-c]pyrrole-2-carboxylic acid tert-butyl ester in overall yield of 35% was obtainted by debenzylation using Pd(OH)2/C as the catalyst.The structures were confirmed by1H NMR,IR and MS.
octahydropyrrolo[3,4-c]pyrrole;azomethine ylide;cycloaddition;synthesis
2013-06-13;
2014-05-28
朱思兰(1988-),女,汉族,浙江金华人,硕士研究生,主要从事有机化学及药物合成的研究。
史海健,教授,硕士生导师,E-mail: shihj@njtech.edu.cn
O626;O621.3
A
1005-1511(2014)05-0664-04
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
民用飞机设计与研究(2020年1期)2020-05-21 07:24:52
中国塑料(2017年2期)2017-05-17 06:13:21
合成化学(2015年10期)2016-01-17 08:56:06
合成化学(2015年10期)2016-01-17 08:55:42
合成化学(2015年1期)2016-01-17 08:53:55
中国塑料(2015年6期)2015-11-13 03:02:49
中国洗涤用品工业(2015年9期)2015-02-28 19:03:05
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
