
新型吡咯季铵盐的合成*
2014-08-30 09:20王玲婷陈衍成郭超伦朱琰婷王世范海南大学海洋学院制药工程系海南海口5708齐鲁制药海南有限公司海南海口57034
合成化学 2014年6期
王玲婷,陈衍成,郭超伦,朱琰婷,王世范[.海南大学 海洋学院 制药工程系,海南 海口 5708;.齐鲁制药(海南)有限公司,海南 海口 57034]
·研究论文·
新型吡咯季铵盐的合成*
王玲婷1,陈衍成2,郭超伦1,朱琰婷1,王世范1
[1.海南大学 海洋学院 制药工程系,海南 海口 570228;2.齐鲁制药(海南)有限公司,海南 海口 570314]
以乙酰乙酸乙酯为原料,经缩合、氧化、还原和氯代等反应合成了3-甲基-5-碘甲基-1H-吡咯-2,4-二(甲酸乙酯)(5);5分别与乌洛托品,吡啶和三甲胺经取代反应合成了3个吡咯季铵盐6a~6c,其中5和6a~6c均为新化合物,其结构经1H NMR,IR和ESI-MS表征。
海洋吡咯生物碱;吡咯衍生物;合成
海洋吡咯生物碱是海洋中蕴藏的一类活性天然产物。20世纪60年代以来,大量海洋吡咯生物碱被分离出来[1-3],其中吡咯季铵盐类生物碱Daminin( Chart 1) 因其对神经的保护作用和低细胞毒性而备受关注[4-5]。但是由于受到海洋天然产物含量稀少的限制,绝大多数海洋吡咯生物碱仍然未能得以深入研究。通过人工合成海洋吡咯生物碱及其衍生物、类似物或结构简化物以探讨其活性,可在深入研究的同时,发现结构更简单、活性更好及毒性更低的新化学实体[6-8]。
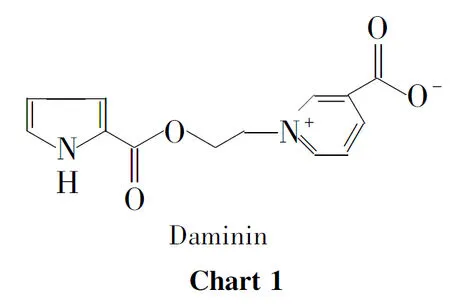
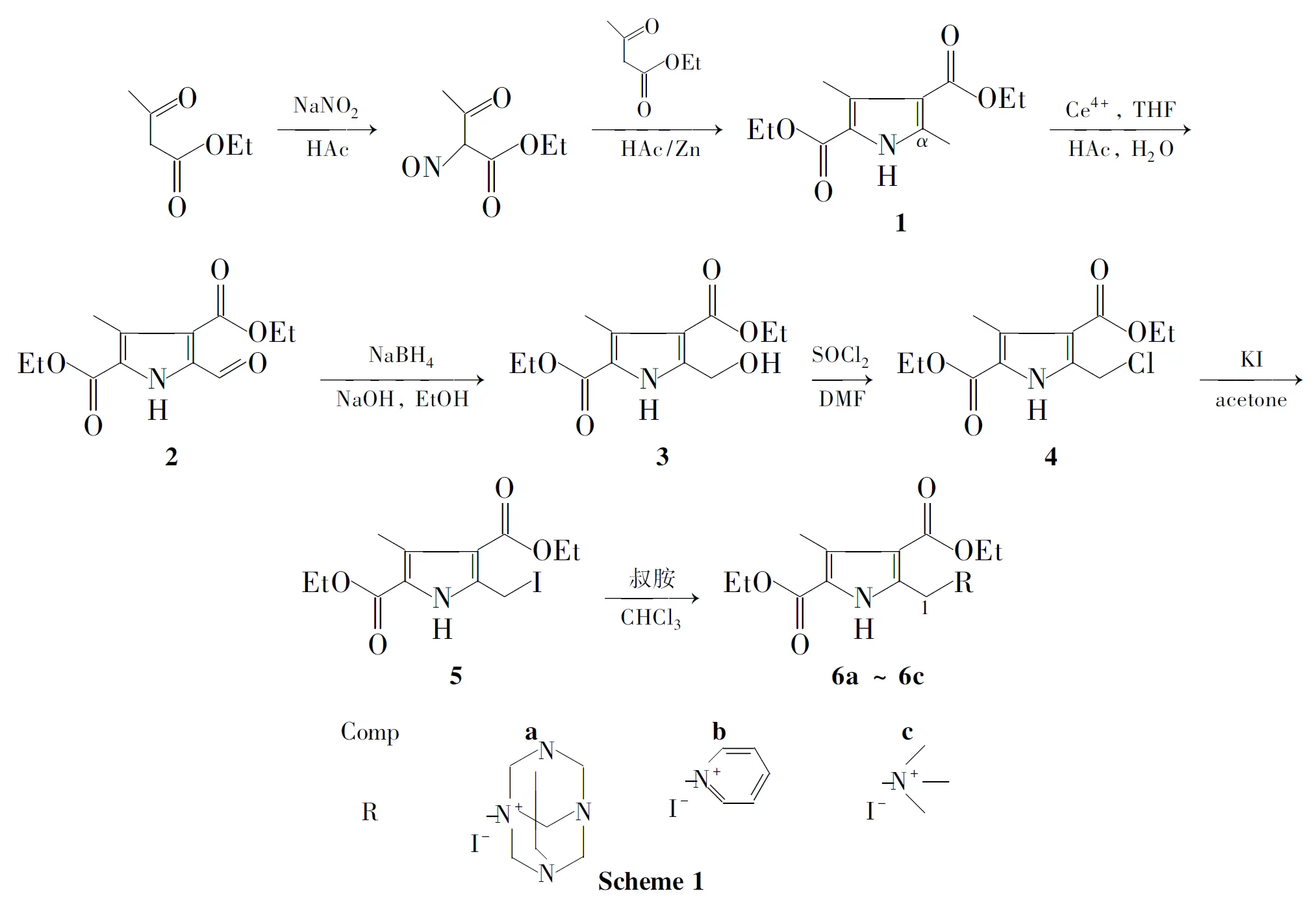
因此,本文以乙酰乙酸乙酯为原料,经缩合、氧化、还原和氯代等反应合成了3-甲基-5-碘甲基-1H-吡咯-2,4-二(甲酸乙酯)(5);5 分别与乌洛托品,吡啶和三甲胺经取代反应合成了3 个吡咯季铵盐6a~6c,其中5和6a~6c均为新化合物,其结构经1H NMR,IR 和ESI-MS 表征。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点仪(温度未校正);Bruker 400MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Thermo iS-10型红外光谱仪(KBr压片);LCMS-IT-TOF型质谱仪。
所用试剂均为分析纯,按标准方法纯化。
1.2 合成
(1)3,5-二甲基-1H-吡咯-2,4-二(甲酸乙酯)(1)的合成[9]
在圆底烧瓶中加入乙酰乙酸乙酯130mL(1.0mol)和冰醋酸500mL,冰水浴降温至<5℃,搅拌下于2h内缓慢滴加NaNO234.5g(0.5mol)的饱和水溶液,滴毕,反应2h(反应液变为无色透明液体)。升温至回流,分别加入锌粉80.0g(1.4mol),加毕,回流反应30min(TLC检测)。冷却至室温,倒入大量冰水中,析出白色絮状固体,搅拌,抽滤,滤饼用水洗涤,干燥得白色絮状固体1,收率80%,m.p.134℃~136℃;1H NMRδ:1.34(t,6H,CH3in Et),2.51(s,3H,3-CH3),2.56(s,3H,5-CH3),4.26(q,2H,CH2in Et),4.30(q,2H,CH2in Et),8.97(s,1H,NH);IRν: 3443,3268,2932,1670,1436,1379cm-1。
(2)3-甲基-5-甲酰基-1H-吡咯-2,4-二(甲酸乙酯)(2)的合成[10]
在反应瓶中加入12.4g(10mmol)和THF 60mL,搅拌使其溶解;加入HAc 60mL和蒸馏水60mL,于室温下搅拌均匀,缓慢加入硝酸铈铵22.0g(40mmol),溶液呈橙黄色,继续反应1.5h(TLC检测)。倒入大量冰水中,析出沉淀,抽滤,滤饼用水洗涤,干燥得白色固体2,收率70%,m.p.124℃~126℃;1H NMRδ: 1.38(t,6H,CH3in Et),2.60(s,3H,3-CH3),4.36(q,2H,CH2in Et),4.39(q,2H,CH2in Et),7.26(s,1H,CHO),10.27(s,1H,NH);IRν: 3444,3270,2932,2899,1699,1478,1261,1086cm-1。
(3)3-甲基-5-羟甲基-1H-吡咯-2,4-二(甲酸乙酯)(3)的合成[11]
在反应瓶中加入22.5g(10mmol)和无水乙醇350mL,搅拌使其溶解;依次加入NaOH 400mg(10mmol)和NaBH4760mg(20mmol),于室温反应1h(TLC检测)。减压蒸除乙醇,倒入水中,析出白色固体,抽滤,滤饼用水洗涤,干燥得白色固体3,收率73%,m.p.121℃~122℃;1H NMRδ:1.36(t,6H,CH3in Et),2.57(s,3H,3-CH3),3.11(s,1H,OH),4.29(m,4H,CH2in Et),4.86(s,2H,1-H),9.24(s,1H,NH);IRν: 3321,3278,2983,2935,1688,1484,1278,1240,1091,778cm-1。
(4)3-甲基-5-氯甲基-1H-吡咯-2,4-二(甲酸乙酯)(4)的合成[11]
在反应瓶中加入32.55g(10mmol)和DMF 15mL,搅拌使其溶解;30min内滴加混合溶液[V(SOCl2)∶V(DMF)=1∶1]10mL,滴毕,于室温反应3.5h至终点(TLC检测)。倒入水中,析出沉淀,抽滤,滤饼用水洗涤,干燥得白色针状晶体4,收率88%,m.p.154℃~156℃;1H NMRδ:1.26(t,6H,CH3in Et),2.58(s,3H,3-CH3),4.25(m,4H,CH2in Et),4.80(s,2H,α-H),9.36(s,1H,NH);IRν: 3244,2985,1674,1480,1280,1202,1093,776cm-1。
(5)3-甲基-5-碘甲基-1H-吡咯-2,4-二(甲酸乙酯)(5)的合成
在反应瓶中加入41.64g(6mmol)和丙酮50mL,搅拌使其溶解;加入KI 4.44g(26.7mmol),于65℃反应4h。倒入水中,析出沉淀,抽滤,滤饼用水洗涤,干燥得淡黄色固体5,收率75%,m.p.176℃~178℃;1H NMRδ:1.25(t,6H,CH3in Et),2.56(s,3H,3-CH3),4.27(m,4H,CH2in Et),4.78(s,2H,1-H),9.38(s,1H,NH);IRν: 3270,2988,1660,1486,1290,1211,1082,790cm-1。
(6)碘化1-{[3,5-二(乙氧羰基)-4-甲基-1H-吡咯-2-基]甲基}-1,3,5,7-四氮杂-1-金刚烷盐(6a)的合成
在反应瓶中依次加入5360mg(1.0mmol),乌洛托品210mg(1.5mmol)和干燥氯仿30mL,加热下搅拌使其溶解,于常温密封静置72h,析出无色针状晶体,干燥得6a328mg,收率65%,m.p.144℃~145℃;1H NMRδ:1.39(m,6H,2CH3),2.59(s,3H,CH3),4.31(m,4H,2CH2),4.56(s,6H,NCH2N),5.38(s,6H,NCH2N),5.39(s,2H,α-H),12.03(s,1H,NH);IRν: 2982,2922,1693,1478,1274,1189,1093,750cm-1;ESI-MSm/z: Calcd for C18H28N5O4I{[M+I]-}632.0236,found 632.0236。
(7)碘化1-{[3,5-二(乙氧羰基)-3-甲基-1H-吡咯-2-基]甲基}-1-吡啶盐(6b)的合成
在反应瓶中加入5360mg(1.0mmol)和干燥氯仿15mL,搅拌使其溶解;滴加无水吡啶0.08mL(1.0mmol),滴毕,加热下搅拌均匀,于室温密封静置72h,析出无色块状晶体,干燥得无色块状固体6b,收率70%,m.p.168℃~170℃;1H NMRδ:1.35(m,6H,CH3in Et),2.55(s,3H,3-CH3),4.28(m,4H,CH2in Et),6.54(s,2H,1-H),8.01(dd,J=7.6Hz,6.8Hz,2H,ArH),8.45(t,1H,ArH),9.61(d,J=6.0Hz,2H,ArH),12.26(s,1H,NH)。IRν: 3131,3022,2988,1714,1683,1568,1488,1275,1240,1080,783,672cm-1;ESI-MSm/z: Calcd for C17H21N2O4I{[M+I]-}570.9596,found 570.9598。
(8)碘化1-[3,5-二(乙氧羰基)-4-甲基-1H-吡咯-2-基]-N,N,N-三甲基甲胺盐(6c)的合成
在反应瓶中依次加入三甲胺盐酸盐20mg(0.2mmol),无水碳酸钾56mg(0.4mmol)和无水氯仿3mL,搅拌下于室温反应10min。过滤得澄清溶液;滴加536mg(0.1mmol)的氯仿(5mL)溶液,滴毕,于室温密封静置72h,自然挥发氯仿,有无色针状晶体析出,过滤,滤饼用乙醇洗涤多次,干燥得无色针状晶体6c,产率50%,m.p.156℃~158℃;1H NMRδ:1.37(m,6H,CH3in Et),2.58(s,3H,3-CH3),3.38(s,9H,3NCH3),4.30(m,4H,CH2in Et),5.44(s,2H,5-CH2N),12.12(s,1H,NH);IRν: 3170,2961,1703,1463,1271,1215,1080,1003,783,650cm-1;ESI-MSm/z: Calcd for C15H25N2O4I{[M+I]-}550.9909,found 550.9907。
2 结果与讨论
2.11的合成
1的合成经过亚硝化和环化两步反应,两步反应需要的反应温度恰好相反。亚硝化反应需要在低温下进行,反应温度一般低于5℃,温度过高,反应产物颜色加深,副反应增加,反应安全性下降。亚硝化反应生成的乙酰乙酸乙酯亚硝化产物,经锌粉还原为胺后与反应体系中的乙酰乙酸乙酯经原位缩合和关环反应生成吡咯衍生物所需温度较高。文献[9]方法采用于室温下一次性加入锌粉后再回流反应,收率71%。本文则在还原反应发生前,先加热反应体系接近沸腾,然后分批加入锌粉,这样有利于吡咯的环化反应发生,避免了锌粉对羰基的还原,收率提高至80%。由于该反应属于放热反应,可利用反应释放的热量维持体系的热平衡。
2.22的合成
吡咯3-位的甲基容易被硝酸铈铵氧化成醛基,文献[10]方法中氧化剂和原料的摩尔比为1∶2,2需经硅胶柱层析纯化,收率仅20%。该反应属于自由基反应,当吡咯α-位上含有吸电子基时,能够稳定反应过程中生成的自由基中间体,使反应容易进行。通过计算,1mol的原料需要4mol的硝酸铈铵才能将甲基全部氧化为醛基,而且介质的酸度对反应很敏感。因此,我们对该反应进行了改进,氧化剂的投料比从1∶2改为1∶4,反应在THF-HAc-H2O混合溶剂的弱酸性体系中进行,反应时间由2h缩短至1.5h,产物通过加入冰水即可获得,收率由20%提高至70%,并且可避免复杂的柱层析纯化。
2.33和4的合成
在3和4的合成中,文献[10]方法采用活泼的吡咯α-位甲基的氯代制得4;4再水解为3的路线。本文则首先将吡咯α-位甲基氧化为醛基,再由NaBH4还原醛基制得3,最后经醇羟基氯代反应合成4。该反应路线不但能够合成3和4,而且能够同时制得中间体2,为多样性导向合成提供了更为丰富的合成模块。
2.46a~6c的合成
碘代烷的反应活性和生物活性远远高于氯代烷。因此,在制备吡咯季铵盐时,先将氯代吡咯用碘化钾置换出碘代吡咯,再与叔胺反应得到碘化吡咯季铵盐6。由于氮原子强的亲核性和碘代烷的活泼性,反应在室温下即可进行。考虑到反应物和产物的溶解性能以及产物的后处理,该反应溶剂选择氯仿较为合适。氯仿不但能够很好地溶解反应物碘代吡咯和叔胺,使反应形成均一体系,而且对6a~6c的溶解性能较差,便于将其从反应体系中分离。
3 结论
利用价廉易得的乙酰乙酸乙酯为原料,经缩合、氧化、还原、氯代等反应高产率地合成了8个吡咯衍生物。1~5可作为基础合成模块,进一步构建更加复杂的吡咯生物碱;新化合物5,6a~6c的生物活性有待进一步研究。
[1] Cafieri F,Fattorusso E,Mangoni A,etal.A novel bromopyrrole alkaloid from the sponge agelas longissima with antiserotonergic activity[J].Bioorg &Med Chem Lett,1995,5(8):799-804
[2] Li X,Ping Huang,Cui J J,etal.Novel pyrrolyllactone and pyrrolyllactam indolinones as potent cyclin-dependent kinase 2inhibitors[J].Bioorg &Med Chem Lett,2003,13(11):1939-1942
[3] Meijer L,Thunnissen A M W H,White A W,etal.Inhibition of cyclin-dependent kinases,GSK-3βand CK1by hymenialdisine,a marine sponge constituent[J].Chemistry &biology,2000,7(1):51-63
[4] Aiello A,D’Esposito M,Fattorusso E,etal.Daminin,a bioactive pyrrole alkaloid from the mediterranean sponge axinella damicornis[J].Tetrahedron,2005,61(30):7266-7270
[5] Bringmann G,Lang G,Tsuruta H,etal.New pyrrole alkaloid daminin and its derivatives,are intracellular calcium ion concentration reducing neuroprotective agents useful for treating neurodegenerative diseases,e.g.Alzheimer’s diseas[P].DE 102004002885,2005
[6] Hoffmanna H,Lindel T.Synthesis of the pyrrole-imidazole alkaloids[J].Synthesis,2003,12:1753-1783
[7] Berrée F,Girard Le Bleis P,Carboni B.Synthesis of the marine sponge alkaloid oroidin and its analogues via suzuki cross-coupling reactions[J].Tetra Lett,2002,43(28):4935-4938
[8] Pla D,Marchal A,Olsen C A,Francesch A,etal.Synthesis and structure-activity relationship study of potent cytotoxic analogues of the marine alkaloid lamellarin D[J].J Med Chem,2006,49(11):3257-3268
[9] Idhayadhulla A,Kumar R S,Nasser A J A,etal.Synthesis of some pyrrole derivatives and their anticoagulant activity[J].American Journal of Drug Discovery and Development,2012,2(1):40-49
[10] Bell I M,Stirdivant S M,Ahern J,etal.Biochemical and structural characterization of a novel class of inhibitors of the type 1insulin-like growth factor and insulin receptor kinases[J].Biochemistry,2005,44(27):9430-9440
[11] Corwin A H,Bailey W A,Viohl J P.Structural investigations upon a substituted dipyrrylmethane:An unusual melting point-symmetry relationship[J].Journal of the American Chemical Society,1942,64(6):1267-1273.
SynthesisofNovelPyrroleQuaternaryAmmoniumSalts
WANG Ling-ting1,CHEN Yan-cheng2,GUO Chao-lun1,ZHU Yan-ting1,WANG Shi-fan1
[1.Department of Pharmaceutical Engineering,School of Ocean,Hainan University,Haikou 570228,China;2.Qilu Pharmaceutical(Hainan)Company Limited,Haikou 570314,China]
An intermediate,diethyl 5-(iodomethyl)-3-methyl-1H-pyrrole-2,4-dicarboxylate(5),was prepared by the reaction of condensation,oxidation,reduction and chlorination from ethyl acetoacetate.Three pyrrole quaternary ammonium salts(6a~6c)was synthesized by substitution reaction of5with urotropin,pyridine and trimethylamine,respectively.5and6a~6cwere novel compunds.The structures were characterized by1H NMR,IR and ESI-MS.
marine pyrrole alkaloid;pyrrole derivative;synthesis
2013-09-09;
2014-09-17
国家自然科学基金资助项目(21162006)
王玲婷(1988-),女,汉族,黑龙江方正人,硕士研究生,主要从事药物合成的研究。
王世范,教授,博士生导师,E-mail:wangsf777@gmail.com
O626.13;O629.3
A
1005-1511(2014)06-0763-04
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
今日农业(2019年11期)2019-08-13
价值工程(2017年31期)2018-01-17
中国司法鉴定(2017年5期)2017-10-11
合成化学(2015年10期)2016-01-17
广州大学学报(自然科学版)(2015年4期)2015-12-23
环境科技(2015年2期)2015-11-08
华东师范大学学报(自然科学版)(2014年4期)2014-03-11
无机化学学报(2014年4期)2014-02-28
中国洗涤用品工业(2012年4期)2012-03-20
