
PI3K-mTOR双重小分子抑制剂的研究进展
2014-08-06 06:52韩进松王重庆宋云龙周永刚第二军医大学药学院药物化学教研室上海00433解放军8医院药剂科江苏南京000
药学实践杂志 2014年5期
陈 颖,韩进松,王重庆,宋云龙,周永刚,朱 驹 (.第二军医大学药学院药物化学教研室,上海 00433; .解放军8医院药剂科,江苏 南京 000)
磷脂酰肌醇3激酶(phosphoinositide 3-kinases, PI3K)是脂激酶家族成员之一,其介导的信号通路在细胞生长、增殖、存活、凋亡和蛋白质的转录,翻译及血管生成中发挥重要的调控作用[1]。PI3K通路与肿瘤的发生、发展、转归有密切关系。该通路已成为近年来广泛研究的药物靶点之一[2,3],尤其在近几年内,许多PI3K小分子抑制剂进入临床研究,其中磷脂酰肌醇3激酶-哺乳动物雷帕霉素靶蛋白(PI3K-mTOR)双重抑制剂的研究成为该领域内的研究热点之一。因此,本文就11 个PI3K-mTOR双重抑制剂的构效关系及研究进展进行综述。
1 PI3K的结构和分类
PI3K是一类复杂的进化保守的酶家族,在哺乳动物细胞内,PI3K根据其结构和生物功能主要分为3型。I型是受体调控的4,5-二磷酸磷脂酰肌醇[PtdIns(4,5)P2]激酶,可分为IA和IB两个亚型。IA亚型(PI3Kα, PI3Kβ和PI3Kδ)是由调节亚基p85和催化亚基p110组成的异质二聚体。IB亚型即PI3Kγ,是由p110γ催化亚基和p101调节亚基构成的异质二聚体[4]。IA和IB两个亚型分别被受体酪氨酸激酶(RTKs)和G蛋白偶联受体(GPCRs)激活。
II型PI3Ks的C端为C2区域,故称其为PI3K-C2激酶,有3种亚型:PI3K-C2α、PI3K-C2β和 PI3K-C2γ,目前关于PI3K-C2s在体内的催化底物还不是很明确,但有研究表明Ⅱ型PI3Ks可以生成3-磷酸磷脂酰肌醇[PtdIns(3)P],也有可能生成3,4-二磷酸磷脂酰肌醇[PtdIns(3,4)P2][5,6]。Ⅲ型PI3Ks由具有催化性的酶Vps34(Vacuolar protein sorting-associated protein 34)和p150亚基构成的异质二聚体组成。虽然Vps34与p110α具有同源性,但是其只利用磷脂酰肌醇(PtdIns)作为底物,而非PtdIns(4,5)P2,即具有磷脂酰肌醇特异性的PI3K活性。此外,有些研究中将PI3K相关蛋白激酶PIKK(其中包括哺乳动物雷帕霉素靶蛋白,mTOR)划分为Ⅳ型PI3K[6]。目前,PI3K的小分子抑制剂的研究主要集中于Ⅰ型PI3K。
2 PI3K信号通路
PI3K-AKT-mTOR信号通路可以被多种途径激活,包括酪氨酸激酶受体如表皮生长因子受体(EGFR),胰岛素样生长因子受体(IGFR),G蛋白偶联受体(GPCRs)。激活后的PI3K催化肌醇在D3位的磷酸化,磷酸化的产物3,4,5-三磷酸磷脂酰肌醇[PtdIns(3,4,5) P3, PIP3]在磷脂酰肌醇依赖性激酶(PDK-1)的作用下直接与蛋白激酶B(AKT)的PH结构域结合,促进AKT的Thr308的磷酸化,在AKT的活化中,Thr308的磷酸化十分关键,但8年后发现哺乳动物雷帕霉素靶蛋白复合物2(mTORC2)能使AKT的Ser473磷酸化,Thr308和Ser473的磷酸化是AKT活化的必要条件。
现已发现有100多种激活后的AKT底物,包括胰岛素信号通路中的糖原合成激酶3(glycogen synthase kinase 3,GSK3),细胞周期调节因子p21以及叉头转录因子(FKHR),同时也能磷酸化BAD,使BCL-2或BCL-XL 被释放,恢复其抗凋亡功能,促进细胞存活。其中mTOR是AKT的重要底物之一,具有丝氨酸/苏氨酸(Ser/Thr)蛋白激酶活性。AKT激活mTOR是通过直接磷酸化mTOR,或者使AKT磷酸化后形成的结节性复合物2(tuberous sclerosis complex 2, TSC2)失活,达到mTOR的激活。mTORC1的下游效应子主要是核糖体p70S6激酶蛋白(S6R1, Kibosomal p70S61 kinase)和翻译抑制因子4EBP1,有调节蛋白质翻译和提高细胞增殖、存活及促血管生成的作用[7]。
3 PI3K的生物学作用
PI3K的小分子抑制剂的研究主要集中于I型PI3K,源于I型PI3K/AKT/mTOR通路在多种肿瘤中的失调。该通路通过PI3K的负调控因子蛋白磷酸酶(phosphatase and tensin homologue deleted on chromosome, PTEN)来调节和抑制PIP2向PIP3的转化。在恶性肿瘤中,常见PTEN的缺失或突变[8]。这些变异使细胞对生长因子的依赖减小,细胞凋亡变慢,促进肿瘤细胞的侵袭。同时编码p110α亚基
的基因PIK3CA过表达[9]。此外,几乎所有的I型PI3K都可由于p85调节亚基的突变而被激活。因而,PI3K通路的抑制成为近年来肿瘤治疗领域非常有前景的治疗策略。
4 PI3K抑制剂的研究进展
1957年,第一个PI3K抑制剂渥曼青霉素从真菌中提取出来,该化合物同时也显示出对mTOR有一定的抑制作用。20世纪90年代Lilly合成了PI3K抑制剂LY294002(图1,对于PI3Ks的IC503.8 μmol/L)。但由于其药物代谢动力学性质不佳、毒性大且选择性差,限制了其被进一步开发为临床药物。随着近10年来相关研究的深入,多种类型的PI3K小分子抑制剂进入临床试验,包括I型广谱PI3K抑制剂(可同时抑制下游mTOR)和亚型选择性的PI3K抑制剂。最近研究已经表明,与mTOR抑制剂相比,PI3K-mTOR的双重抑制剂可以通过对该信号通路上的多个关键位点进行抑制,同时防止AKT介导的信号的增加,达到抗肿瘤效果增强的作用。另外,PI3K的p110亚基和mTOR的催化结构域之间的结构相似性极大地推动了该类双重抑制剂的发展[3]。
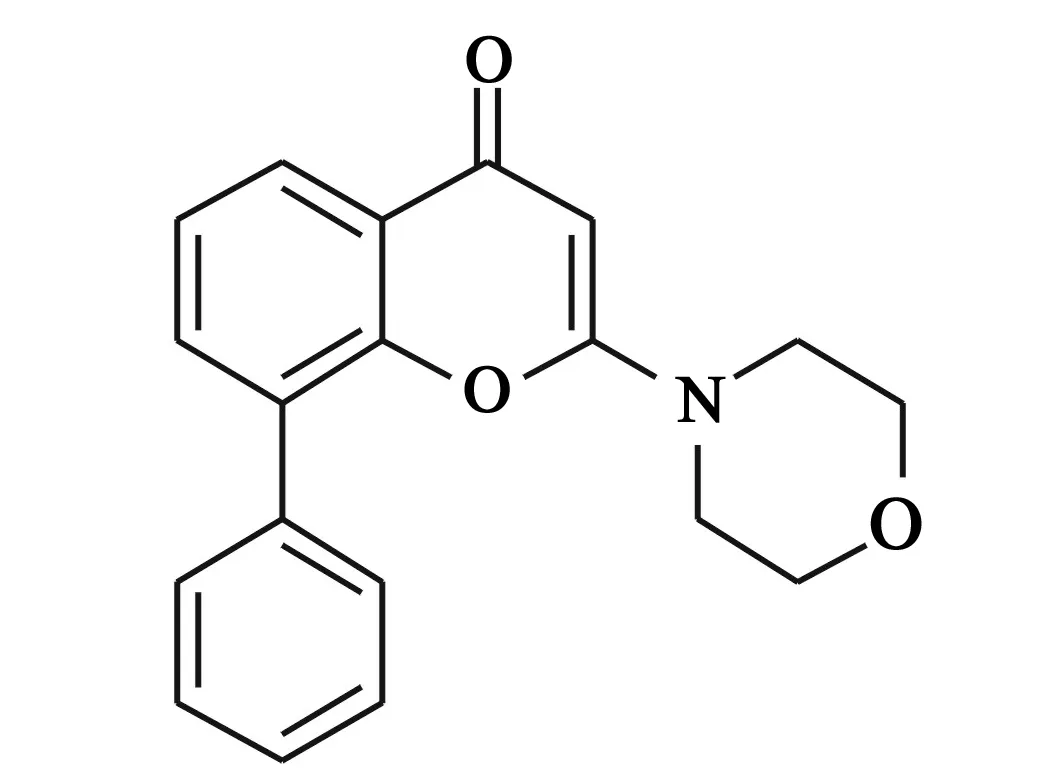
图1 LY294002化学结构式
4.1GDC0980 GDC0980是具有噻吩[3,2-d]嘧啶骨架的吗啉芳基衍生物(结构见图2)。结构与GDC0941很类似,用2-氨基嘧啶替代GDC0941(结构见图2)结构中的吲哚基团后,GDC0980具有PI3K-mTOR双重抑制活性(对于PI3Kα、PI3Kβ、PI3Kδ、PI3Kγ的IC50值分别为5、27、7、14 nmol/L;对于mTOR, Ki 值为17 nmol/L)。
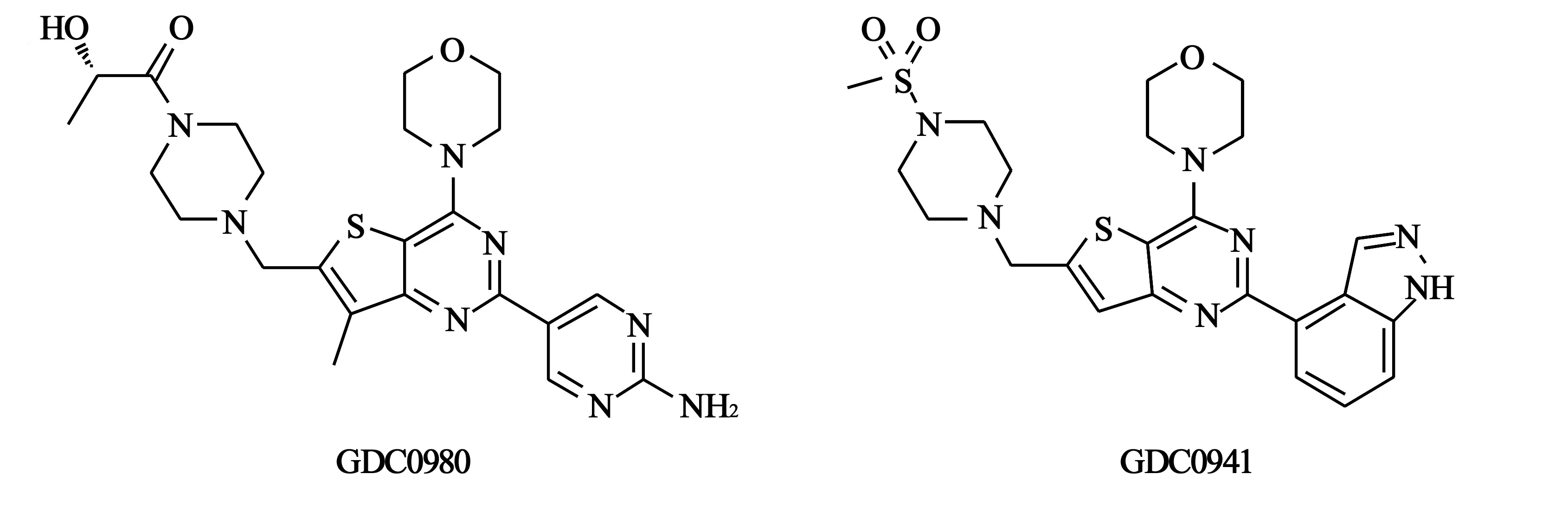
图2 GDC0980和GDC0941化学结构式
4.2XL765 该化合物(结构见图3)对PI3Kα、PI3Kβ、PI3Kδ、PI3Kγ的IC50值分别为39、113、43、9 nmol/L;对mTORC1和mTORC2的IC50值分别为190、908 nmol/L。目前对其构效关系和结合模式的报道较少[11]。
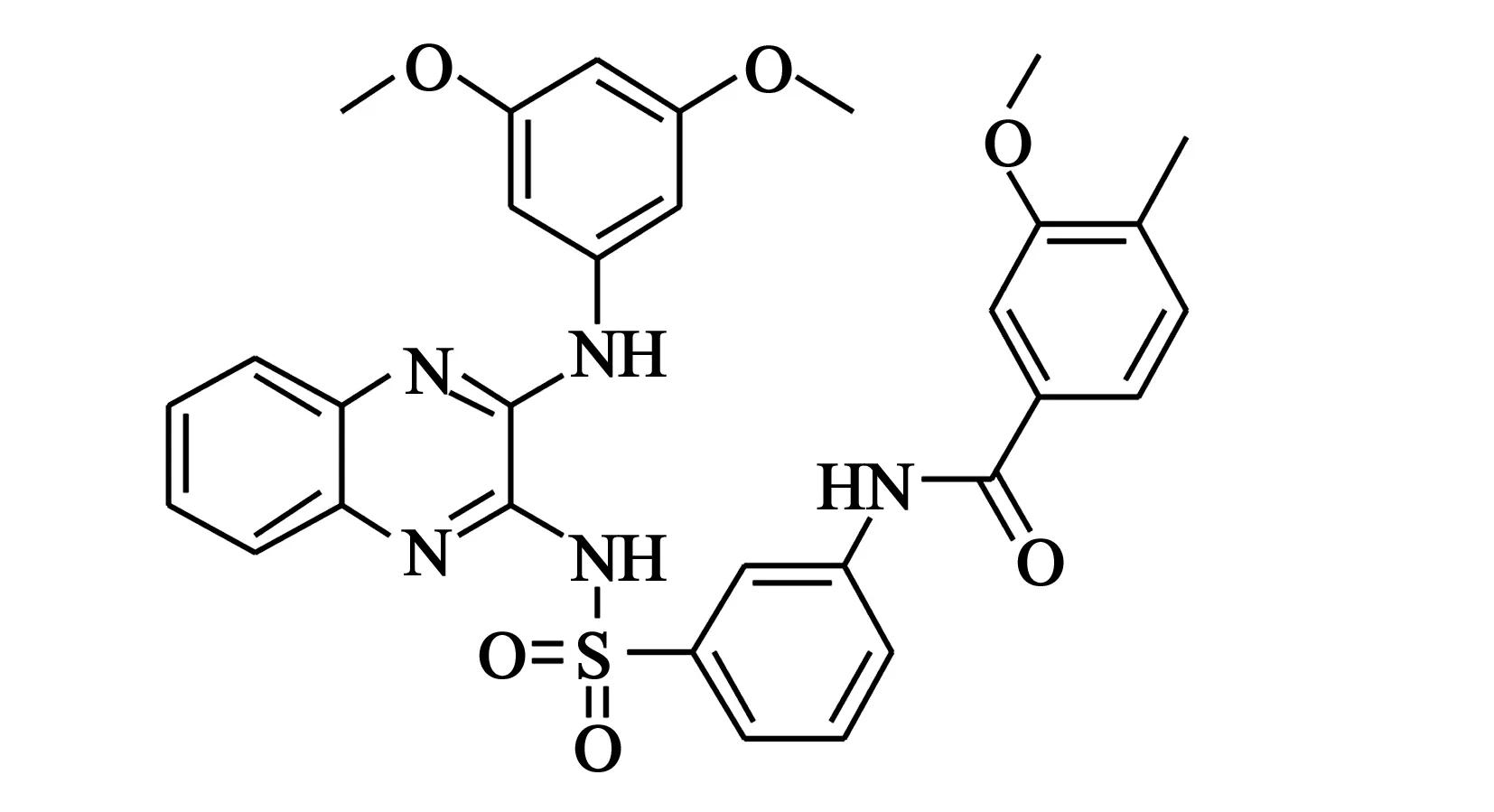
图3 XL765化学结构式
4.3SF1126 在化合物LY294002的基础上研究前药,发现了化合物SF1126(结构见图4)。由于LY294002的选择性较低,可溶性较差,药效较低,故将LY294002与血管靶向的四肽SF1174结合,用于抗癌治疗。SF1126对于PI3Kα、PI3Kβ、PI3Kδ、PI3Kγ以及mTOR的IC50值分别为356、736、3 225、1 774、1 060 nmol/L,能够通过与特异性的整合素结合而聚集在肿瘤新生血管周围,并选择性在肿瘤组织中释放LY294002[12]。LY294002的水溶性较差,半衰期较短,而SF1174水溶性好,并显示出较好的药代动力学特性,且该化合物在多种动物模型中具有较好的耐受性,对多种人类肿瘤具有较强的体内抑瘤活性[13]。
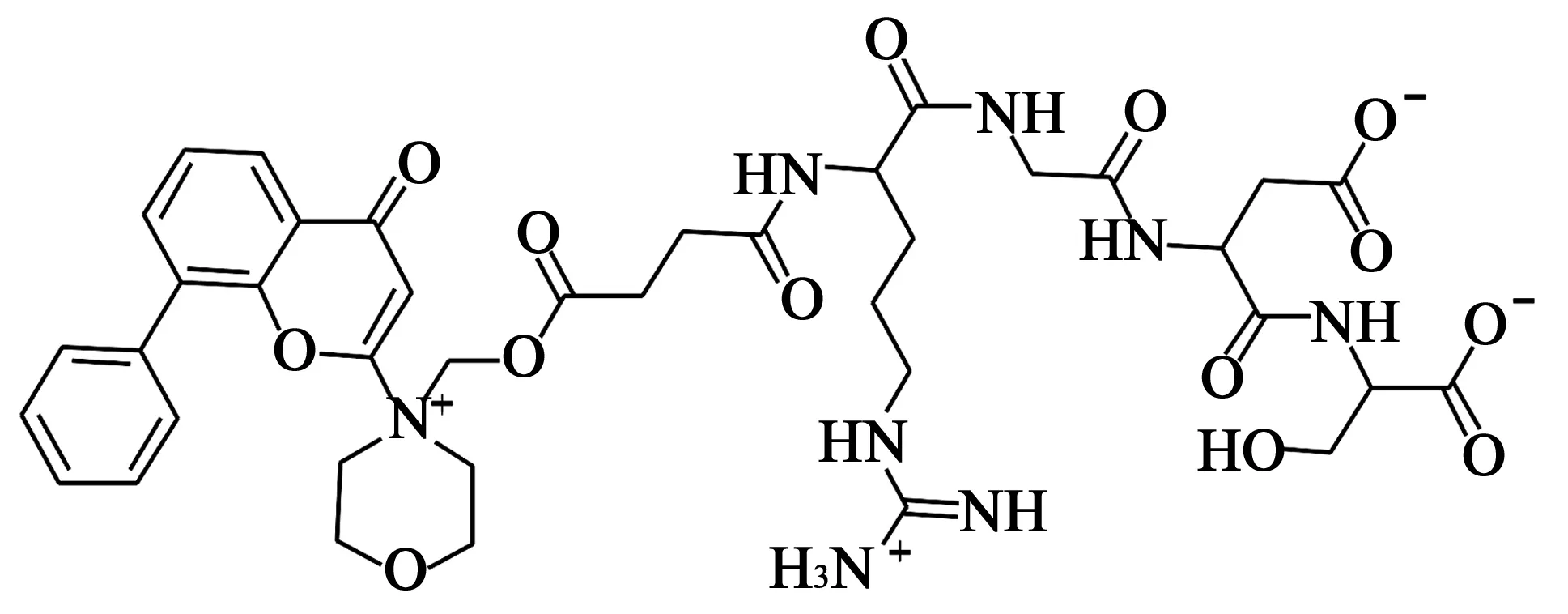
图4 SF1126化学结构式
4.4BEZ235 该化合物是以咪唑并[4,5-c]喹啉酮为骨架的双重抑制剂(结构见图5),对PI3Kα、PI3Kβ、PI3Kδ、PI3Kγ以及mTOR的IC50值分别为4、75、 7、5、21 nmol/L,能有效抑制突变的PI3Kα。对于VEGFR、 c-Src、 PKA、AKT和PDK1等一系列蛋白激酶活性较差[14]。另外,BEZ235对恶性胶质母细胞瘤(U87MG)和PC3M肿瘤异种移植有显著的抑瘤活性。以咪唑喹啉为骨架的先导化合物通过以结构为基础的设计方法得到BEZ235,以期模仿与ATP的腺嘌呤部分的氢键结合[14]。利用同源模建PI3Kα来进行对接研究,结果显示,BEZ235与Val851、Asp933、Ser774之间存在复杂的氢键结合,与ATP结合位点的一些保守的疏水残基之间存在范德华力。通过mTOR同源模建后进行对接,发现BEZ235具有类似的结合模式[14]。
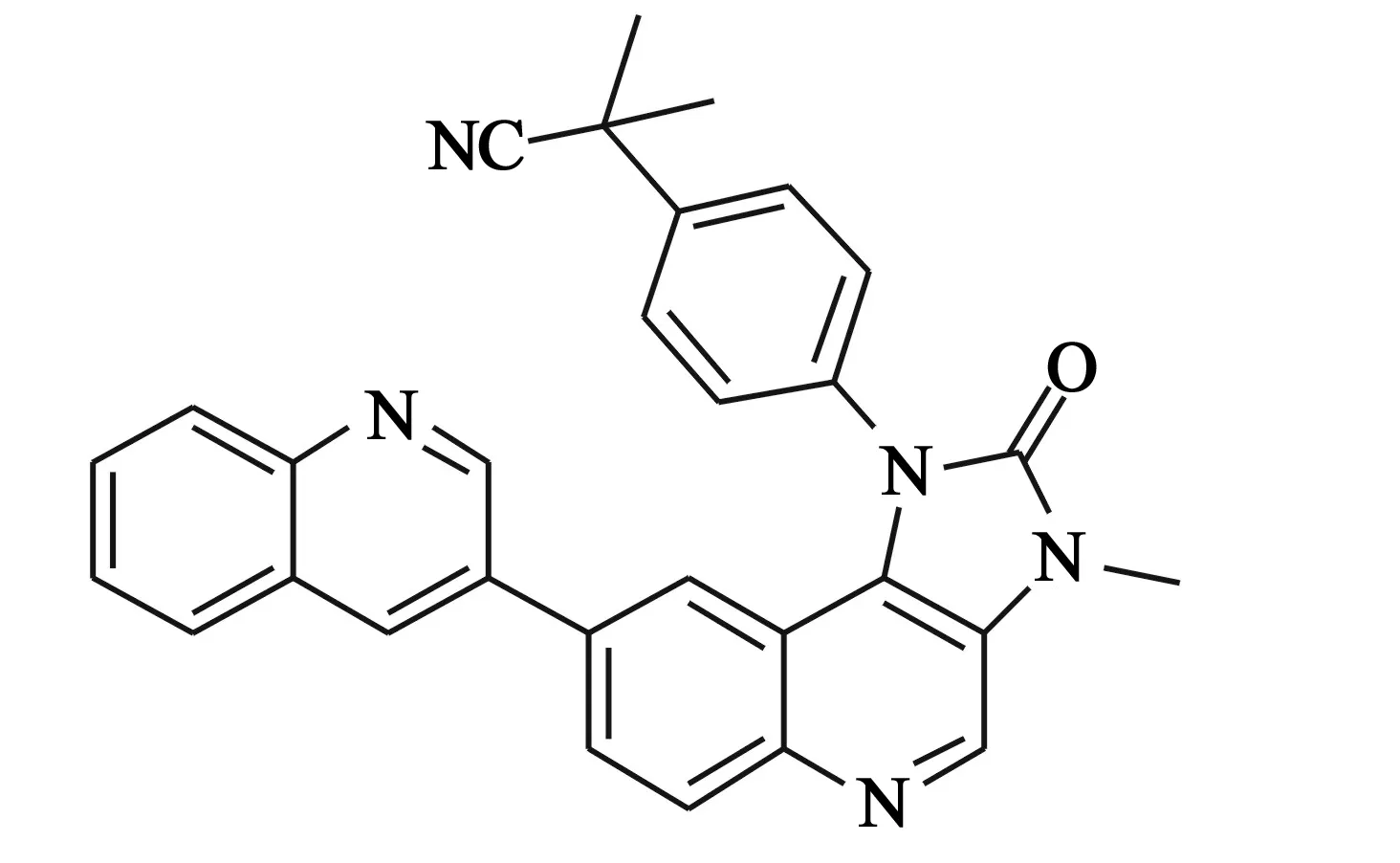
图5 BEZ235化学结构式
4.5PF04691502 该化合物具有优秀的激酶选择性,良好的ADMET性质和体内抗肿瘤活性,对于PI3Kα和mTOR的Ki值分别为0.57和16 nmol/L(结构见图6)。该化合物通过基于结构的设计方法从一系列4-甲基吡啶嘧啶酮类(MPPs)化合物中得到。在这系列MPPs中,PF00271897(结构见图6)因其对PI3Kα的Ki值为1.4 nmol/L,对mTOR的Ki值为840 nmol/L,以及其较高的配体效率(LE=0.48)而被作为进一步物理性质优化筛选的候选化合物。这两个化合物与PI3Kγ的共晶结构显示,两个化合物的氨基嘧啶部分都能与PI3Kγ铰链区的Val882残基形成氢键结合。在晶体复合物中,PF04691502结构中嘧啶环的氮原子与一个结合水分子能在亲水口袋形成水桥,而在PF00271897-PI3Kγ复合物中,苯甲醇基团占据了该位点。同时,这两个化合物的母环吡啶嘧啶酮结构能在铰链区形成疏水口袋。该口袋特异性地存在于PI3Ks和mTOR,与化合物存在激酶选择性有关[10]。
4.6GSK2126458 葛兰素史克公司在化合物GSK1059615的化学结构基础上,利用基于结构的设计方法得到GSK2126458,其具有高活性、高选择性和良好的药动学特性(结构见图7)[15]。该化合物最初的设计策略是将GSK1059615(结构见图7)中的噻唑烷二酮结构替换为更大的片段以便更紧密结合于酶口袋。后续的结构修饰将着眼于提高化合物的水溶性,改善药代动力学特性。通过对该化合物与PI3Kγ的共晶结构分析,可知化合物在生理pH条件下,结构中的磺酰胺的-NH部分与Lys833形成静电作用,这有可能解释了其活性普遍优于已报道的PI3K抑制剂的原因。此外,化合物结构中吡啶环的氮原子通过水分子的桥连作用形成氢键,喹啉环的氮原子与Val882形成氢键,二氟苯基基团在Asp964附近填充疏水区域(其与PI3Kγ的结合模式见图8[15])。

图6 PF00271897和PF04691502化学结构式
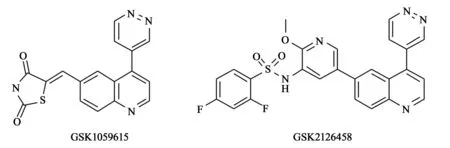
图7 GSK1059615和GSK2126458化学结构式
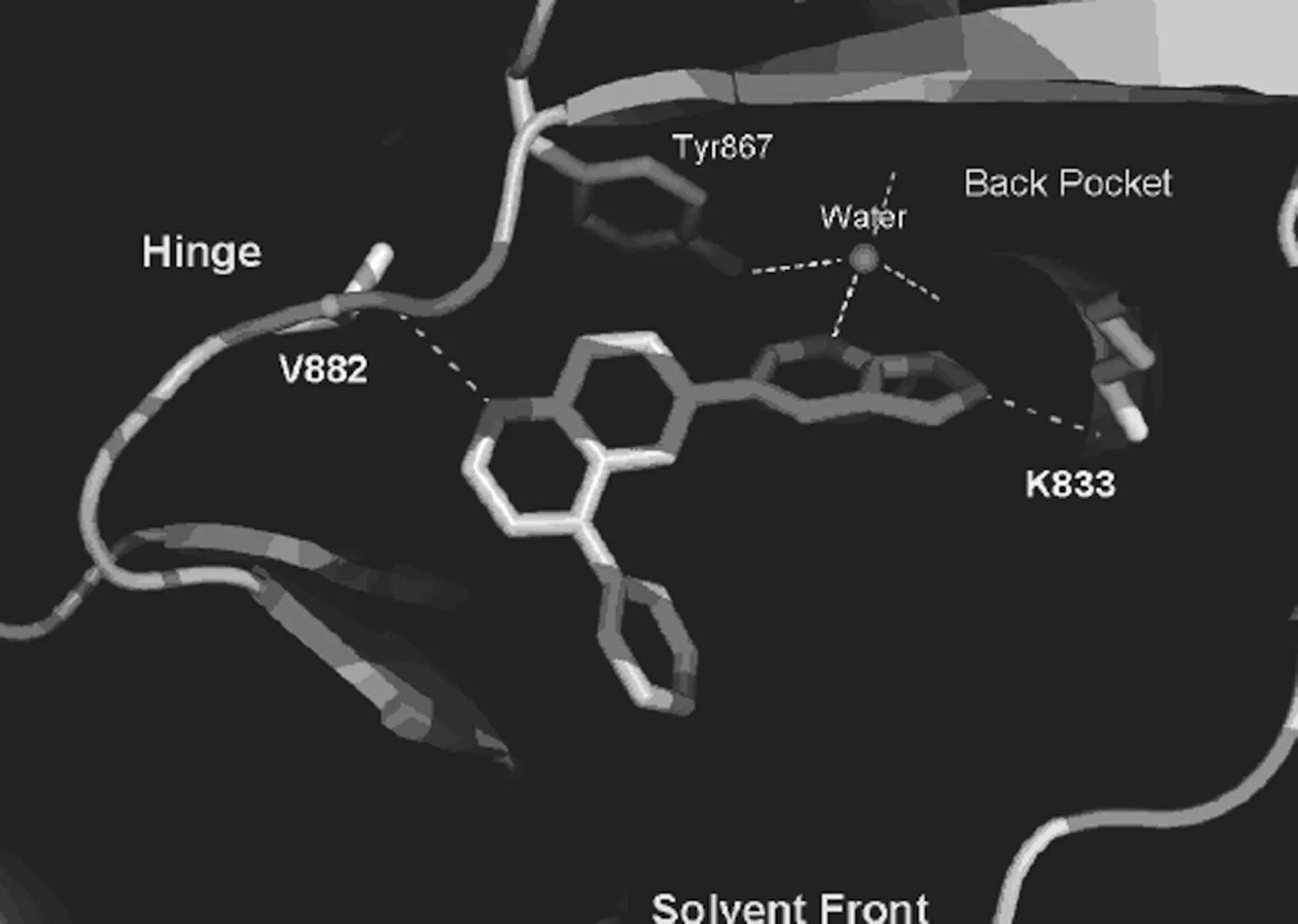
图8 GSK2126458与PI3Kγ的结合模式
该化合物对于常见的突变型PI3Kα显示皮摩尔(pmol)级活性(对于PI3Kα的Glu542、Glu545和His1047突变型的Ki值分别为0.008、0.008、0.009 nmol/L),对mTOR(对mTORC1和mTORC2的Ki值分别为0.18、0.30 nmol/L)之外的蛋白激酶有很高的选择性。该化合物不仅酶活性高,细胞活性也很强,同时体内抗肿瘤活性优异。
4.7PKI587 该化合物是近来出现的另一个PI3K-mTOR双重抑制剂(对PI3Kα、PI3Kγ、mTOR的IC50值分别为0.4、5.4、1.6 nmol/L)(结构见图9)[16]。与先前报道的以三唑并嘧啶为骨架的化合物PKI402 (结构见图9) (对PI3Kα、PI3Kγ、mTOR的IC50值分别为1.4、9.2、1.7 nmol/L)结构相似性很高,将母核中的嘧啶环替换为1,3,5-三嗪,来提高PKI587的溶解度。在化合物中引入两个吗啉基团的目的是即使其中一个吗啉环在体内发生氧化代谢,还有另一个能与铰链区的缬氨酸残基发生氢键相互作用[16,17]。PKI587对于乳腺癌,结肠癌,肺癌及神经胶质瘤的异体移植模型具有显著抗癌活性[18]。
4.8BGT226 该化合物与BEZ235具有相同的咪唑并[4,5-c]喹啉酮骨架(结构见图10)。BGT226能抑制乳腺癌细胞和经放疗后的肿瘤细胞中的Akt和S6的磷酸化。实验表明,该化合物对于若干肿瘤细胞株显示出纳摩尔级的活性。BGT226在临床一期剂量递增研究中,显示出快速吸收,且半衰期在6~9 h[19, 20]。
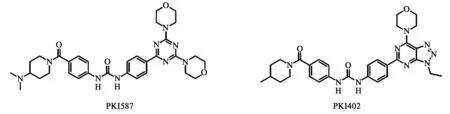
图9 PKI587和PKI402化学结构式
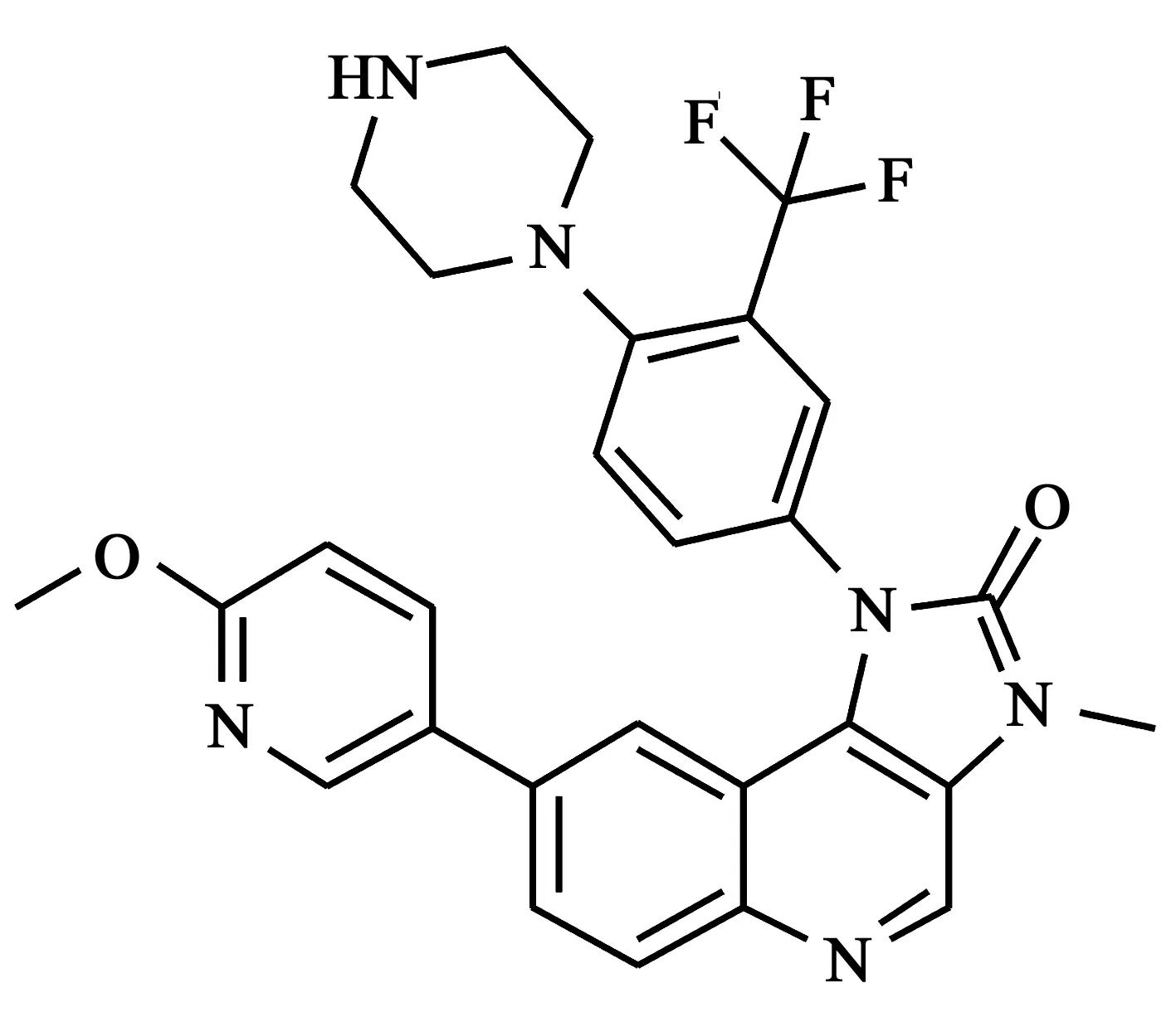
图10 BGT226化学结构式
4.9GNE477 研究人员在对噻吩嘧啶类衍生物GDC0941进行一系列结构修饰时,发现双重抑制剂GNE477(对PI3Kα的IC50值为2 nmol/L,对mTOR的Ki值为29 nmol/L),对肿瘤细胞PC3的活性优良,药代动力学性质理想(结构见图11)。构效关系研究表明,GDC0941结构中的吲哚基团被2-氨基嘧啶替换后活性保持(结构见图11中化合物1),对PI3Kα的IC50值为3 nmol/L),该化合物同时显示出对mTOR的抑制活性(Ki值为29 nmol/L)。在噻吩嘧啶母环的6位无取代时,化合物2 (结构见图11)同样显示出对PI3K-mTOR的双重抑制活性(对PI3Kα的IC50值为5 nmol/L,对mTOR的Ki值为42 nmol/L),由此可知,活性的保持主要是由于吗啉取代的噻吩嘧啶部分的存在。化合物GNE477在化合物1母环7位引入甲基以达到打破平面结构降低熔点以及提高口服生物利用度的目的[21]。
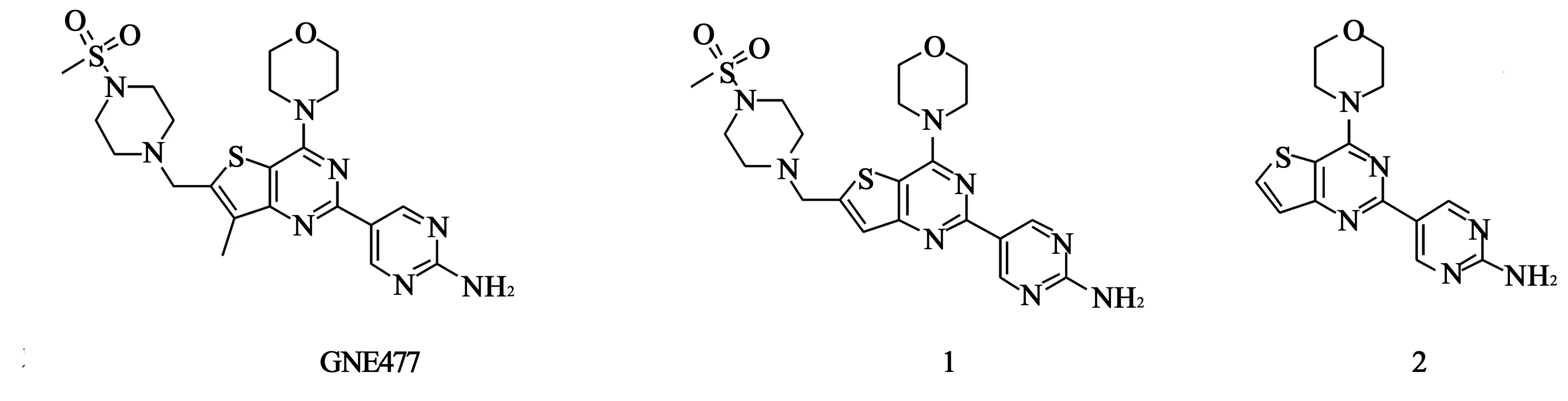
图11 GNE477及化合物1、2化学结构式
4.10PWT33597和DS7423 化合物PWT33597在体外显示出高度的选择性,同时对于肿瘤组织分布较好,通路抑制有效,在多种遗传背景的异体移植肿瘤模型中显示优良的口服有效性。该化合物药代动力学性质良好,正在进行临床Ⅰ期试验。化合物DS7423目前也在进行临床Ⅰ期试验[22]。
5 总结
笔者综述了近年来进入临床研究的PI3K-mTOR双重小分子抑制剂。该通路上的关键靶酶已成为抗肿瘤治疗领域很有前景的药物靶点,对于药物化学工作者来说,既是机遇,也是挑战,如何开发出高活性、药动学性质良好的PI3K-mTOR小分子抑制剂,并正确使用于临床,将有许多难题需要克服。如目前出现的耐药问题以及临床上的联合用药方案都有待进一步研究。
自2006年随着小分子抑制剂BEZ235进入临床研究,接下来的六七年中,10多个很有治疗前景的双重PI3K-mTOR小分子抑制剂陆续进入不同阶段的临床研究。这些研究也揭示了该靶点的一些普遍特征。例如通过对一系列文献报道的化合物分子的晶体复合物结构的分析,笔者发现很多结构类型小分子与铰链区的缬氨酸残基存在氢键作用,与亲水口袋的赖氨酸与天冬氨酸残基形成氢键。这些缔合上的特点,为深入研究新型高效、高选择性的PI3K-mTOR双重小分子抑制剂提供了有效的研究指导。
【参考文献】
[1] Vivanco I, Sawyers CL, The phosphatidylinositol 3,kinase AKT pathway in human cancer[J]. Nat Rev Cancer, 2002,2(7): 489-501.
[2] Bjornsti MA, Houghton PJ, The TOR pathway: a target for cancer therapy[J]. Nat Rev Cancer, 2004,4(5): 335-348.
[3] Workman P, Clarke PA, Raynaud FI,etal.Drugging the PI3 kinome: from chemical tools to drugs in the clinic[J]. Cancer Res, 2010,70(6): 2146-2157.
[4] Denley A, Kang S, Karst U,etal.Oncogenic signaling of class I PI3K isoforms[J]. Oncogene, 2008, 27(18): 2561-2574.
[5] Falasca M, Maffucci T, Role of class II phosphoinositide 3-kinase in cell signalling[J]. Biochem Soc Trans, 2007,35(Pt 2): 211-214.
[6] Guertin DA, Sabatini DM. Defining the role of mTOR in cancer[J]. Cancer Cell, 2007,12(1): 9-22.
[7] Bunney TD, Katan M.Phosphoinositide signalling in cancer: beyond PI3K and PTEN[J]. Nat Rev Cancer, 2010,10(5): 342-352.
[8] Bussink J, van der Kogel AJ, Kaanders JH, Activation of the PI3,K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer[J]. Lancet Oncol, 2008,9(3): 288-296.
[9] Massion PP, Kuo WL, Stokoe D,etal. Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization: implications of the phosphatidylinositol 3-kinase pathway[J]. Cancer Res, 2002,62(13): 3636-3640.
[10] Sutherlin DP, Bao L, Berry M,etal. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer[J]. J Med Chem, 2011,54(21): 7579-7587.
[11] Wu P, Hu YZ.Small molecules targeting phosphoinositide 3-kinases[J]. Med Chem Comm, 2012,3(11): 1337-1355.
[12] Garlich JR, De P, Dey N,etal. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity[J]. Cancer Res, 2008,68(1): 206-215.
[13] Ozbay T, Durden DL, Liu T,etal.Invitroevaluation of pan-PI3-kinase inhibitor SF1126 in trastuzumab-sensitive and trastuzumab-resistant HER2-over-expressing breast cancer cells[J]. Cancer Chemother Pharmacol, 2010,65(4): 697-706.
[14] Stauffer F, Maira SM, Furet P,etal. Imidazo[4,5-c]quinolines as inhibitors of the PI3K/PKB-pathway[J]. Bioorg Med Chem Lett, 2008,18(3): 1027-1030.
[15] Knight SD, Adams ND, Burgess JL,etal. Discovery of GSK2126458, a highly potent Inhibitor of PI3K and the mammalian target of rapamycin[J]. Acs Med Chem Lett, 2010,1(1): 39-43.
[16] Venkatesan AM, Dehnhardt CM, Delos SE,etal. Bis(morpholino-1,3,5-triazine)derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26(PKI-587), a highly efficacious dual Inhibitor[J]. J Med Chem, 2010,53(6): 2636-2645.
[17] Dehnhardt CM, Venkatesan AM, Delos SE,etal. Lead optimization of N-3-substituted 7-morpholinotriazolopyrimidines as dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors: discovery of PKI-402[J]. J Med Chem, 2010,53(2): 798-810.
[18] Mallon R, Feldberg LR, Lucas J,etal.Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor[J]. Clin Cancer Res, 2011,17(10): 3193-3203.
[19] Cheng HM, Bagrodia S, Bailey S,etal. Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04691502 through structure based drug design[J]. Med Chem Comm, 2010,1(2): 139-144.
[20] Markman B, Tabernero J,Krop I,etal. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors[J]. Ann Oncol, 2012,23(9): 2399-2408.
[21] Heffron TP, Berry M, Castanedo G,etal. Identification of GNE-477, a potent and efficacious dual PI3K/mTOR inhibitor[J]. Bioorg Med Chem Lett, 2010,20(8): 2408-2411.
[22] Kashiyama T, Oda K, Ikeda Y,etal.Antitumor efficacy of DS-7423, a novel PI3K/mTOR dual inhibitor, in ovarian clear cell adenocarcinoma[J]. Eur J Cancer, 2012,48: 110-111.
猜你喜欢
中国饲料(2022年6期)2022-04-22
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
宁夏医学杂志(2020年4期)2020-03-01
宁夏医学杂志(2020年3期)2020-02-27
农药科学与管理(2019年6期)2019-11-23
中国饲料(2019年12期)2019-07-12
武警医学(2018年10期)2018-11-06
中学生数理化·高三版(2016年12期)2017-03-02
现代检验医学杂志(2016年1期)2016-11-12
