胰岛β细胞功能衰退之谜?——内质网应激在β细胞功能衰退中的作用
2014-07-26 14:47解放军总医院内分泌科母义明
药品评价 2014年9期
解放军总医院内分泌科 母义明
胰岛素抵抗和胰岛β细胞功能障碍是2型糖尿病(T2DM)的两大发病机制[1]。研究显示,胰岛素抵抗早期即可发生,与非肥胖的正常糖耐量(NGT)者相比,肥胖的NGT者的胰岛素敏感性已经下降了29%,肥胖的葡萄糖耐量异常(IGT)者的胰岛素敏感性进一步下降28%,而β细胞功能衰退的发生及速度则决定了糖尿病的发病及进程[2]。因此,胰岛素抵抗可能是T2DM发生的始动因素,当胰岛β细胞功能不能继续代偿胰岛素生理需求时血糖开始升高,最终进展为糖尿病[3],其中胰岛素的早相分泌丧失发生较早,而各种因素所导致的胰岛β细胞数量减少及功能紊乱是糖尿病致病机制的核心[4]。
胰岛β细胞衰退的揭秘
2013年第49届欧洲糖尿病研究学会(EASD)学术年会上将第48 届Minkowski奖授予来自比利时布鲁塞尔大学(ULB)伊拉斯姆斯医院的Miriam Cnop教授,以表彰她长期致力于β细胞功能基础研究的巨大贡献。
Miriam Cnop教授的研究小组一直关注胰岛β细胞及凋亡在单基因糖尿病及T2DM发病中的作用,阐明了游离脂肪酸(free fatty acid,FFA)诱导β细胞凋亡的信号转导通路,发现内质网(endoplasmic reticulum,ER)应激在FFA诱导的β细胞凋亡中发挥了重要作用,并积极寻找能保护β细胞的治疗靶标及干预策略。
1.T2DM的一级亲属早期即存在胰岛β细胞功能的减退
Cnop等[5]研究在33名T2DM患者的非糖尿病一级亲属中连续7年采用静脉及口服葡萄糖耐量试验观察其胰岛素敏感性(insulin sensitivity,SI)、β细胞功能、葡萄糖自身代谢效应(glucose effectiveness,Sg)及葡萄糖耐量情况,结果显示受试者的体重和腹围虽然逐渐增加,但其SI、葡萄糖刺激后急性胰岛素反应(AIRg)、Sg并未见显著变化。然而当调整了胰岛素敏感性对胰岛素释放的作用后,作为反映β细胞功能的葡萄糖处置指数 [disposition index,DI(=AIRg*SI)]下降了22%(P<0.05)。初始评估为NGT的受试者,在之后的随访中有部分进展为IGT,与未进展为IGT的受试者相比,那些进展为IGT的受试者的DI下降达50%(P<0.05)。因此提示,T2DM的一级亲属的糖耐量减低与β细胞功能减退相关。
2.β细胞功能丧失的关键是β细胞的衰亡增加而并非增生能力下降
与啮齿类动物不同,随着年龄增加,人和哺乳动物的许多组织细胞尤其是那些氧化应激水平较高、寿命较长的组织细胞如神经元细胞、心肌细胞、胰岛β细胞内的脂褐素小体(lipofuscin bodies,LBs))沉积增加。在胰岛细胞,不仅线粒体和内质网膜经自噬作用(autophagy)发生溶酶体酶解退化,而且过量的分泌颗粒及细胞内膜也发生酶解退化,从而使胰岛β细胞内的色素及金属离子在细胞内堆积,形成LBs,因此LBs的沉积代表着废物的贮存并聚集在细胞内而不能运送出细胞外。细胞内LBs沉积的速率、大小及成分与细胞活动如ER应激、线粒体更新、分泌颗粒降解等有关,所以评估脂褐素沉积的量可以作为细胞老化和细胞寿命的标志。人类尸检的电镜标本显示,在生命早期(1岁)时胰岛β细胞内就有脂褐素沉积存在,随年龄增加β细胞LBs沉积量显著增加,胰岛内含有LBs的β细胞比例也有增加。在其他灵长类动物如猴子(5~30岁)的研究中也有相似的结果。但作为啮齿类的小鼠试验的结果则显示细胞内LBs的含量不足人类的10%,只在60周龄以上的小鼠细胞内LBs才有所增加。然而人体内分裂旺盛的细胞,如人类胰岛瘤细胞中的LBs含量也表现较少,且与年龄无关。正常情况下估算,人在20岁时LBs阳性的胰岛β细胞比例可达97%,因此提示分化新生的胰岛β细胞比例极少(不足3%),大部分的胰岛β细胞在20岁以前已经形成并达到高峰,无证据表明20岁以后β细胞还有不断新生和复制。不管是LBs含量还是LBs阳性细胞比例均与BMI或T2DM无关,提示成人胰岛并非以细胞复制来应对胰岛素需求增加。因此认为,β细胞的功能异常和凋亡,而非β再生能力下降是T2DM发病的关键因素[6,7]。综上,正常人和T2DM患者的胰岛β细胞增生都不活跃,早期即可发生的β细胞凋亡和功能异常是T2DM发生、发展的关键。
3.目前公认的导致胰岛β细胞衰退的原因有以下几种:
3.1 脂毒性作用 Cnop等[8]研究显示,人的胰岛β细胞内有富含LDL及vLDL的脂质贮存颗粒(lipid-storing vesicles,LSVs)的聚集。随着增龄,β细胞内LSVs含量逐渐增加,这可能导致β细胞长期暴露于高脂环境下。β细胞内FFA聚集可诱导NO合酶产生及NO介导的β细胞凋亡;FFA增加线粒体的脂质氧化,使线粒体ATP生成减少,代谢偶联因子下降,并从而产生更多的活性氧(ROS),诱导激活自噬溶酶体的酶解作用;FFA还通过诱导β细胞的ER应激引发细胞凋亡。因此,脂毒性促进β细胞衰退,是导致T2DM发病的重要因素[9]。
3.2 葡萄糖毒性作用
研究表明,中、重度血糖增高可以降低机体葡萄糖诱导的胰岛素分泌(glucose-induced insulin secretion,GIIS),促进IGT进展为糖尿病。长期暴露于高糖条件下的大鼠胰岛β细胞的表型发生改变[10]。事实上,长期暴露于高糖条件具有双重作用,一些研究结果表明,β细胞的GIIS缺陷是由于高糖诱导下的ATP合成减少,称之为“对葡萄糖的敏感性下降”;而另一些研究则显示,β细胞暴露于高糖环境使其对高糖的敏感性增高,从而刺激线粒体代谢、胰岛素原的合成及胰岛素的分泌,这将引发在β细胞即使在低浓度葡萄糖时也存在ATP生成过多。因此在长时间暴露于高糖条件下时,虽然部分存活的β细胞仍存在对“葡萄糖的高敏感性”,但同时部分β细胞也已经发生了凋亡[11]。此外,Bachar等[12]研究还发现,高糖通过增高IRE1α蛋白水平及激活JUN途径,可以促进FFA诱导的ER应激,促进β细胞功能紊乱及导致β细胞凋亡的发生。
3.3 氧化应激及线粒体途径
研究表明,GIIS可增强线粒体ATP信号,从而增加胰岛β细胞线粒体的代谢流,而高脂可以增强这个作用。高糖、高脂通过呼吸链增加线粒体代谢流,进而增加活性氧(ROS)的生成;另一方面,随之通过解偶联蛋白2(UCP2)的作用降低线粒体膜的电压,从而降低呼吸链的代谢流和活性氧的生成,发挥对ROS诱导损伤的保护性机制,但同时降低由代谢物质诱导的胰岛素的分泌[13]。β细胞由于缺乏抗氧化酶的表达,尤其易受到ROS攻击;增高的ROS破坏GIIS,降低β细胞关键基因的表达,并诱导细胞凋亡[14]。此外,氧化应激还触发胰岛素抵抗,从而在T2DM的发生发展过程中起重要的作用[15]。3.4 内质网(ER)应激途径
ER应激是在细胞在高分泌活动时的重要适应性细胞信号反应,又称非折叠蛋白反应(unfolded protein response,UPR)。这种细胞反应的目的是通过降低蛋白质翻译,上调ER的分子伴侣(因此增加ER折叠能力)以及降解错误折叠的蛋白从而恢复ER稳态[16]。当错误折叠的蛋白质聚集在内质网腔内引发ER应激时,导致ER膜蛋白IRE1、ATF6及PERK活化,这些ER应激转换蛋白与ER分子伴侣(BiP)解离而激活,导致细胞内信号的转导[17](见图1)。
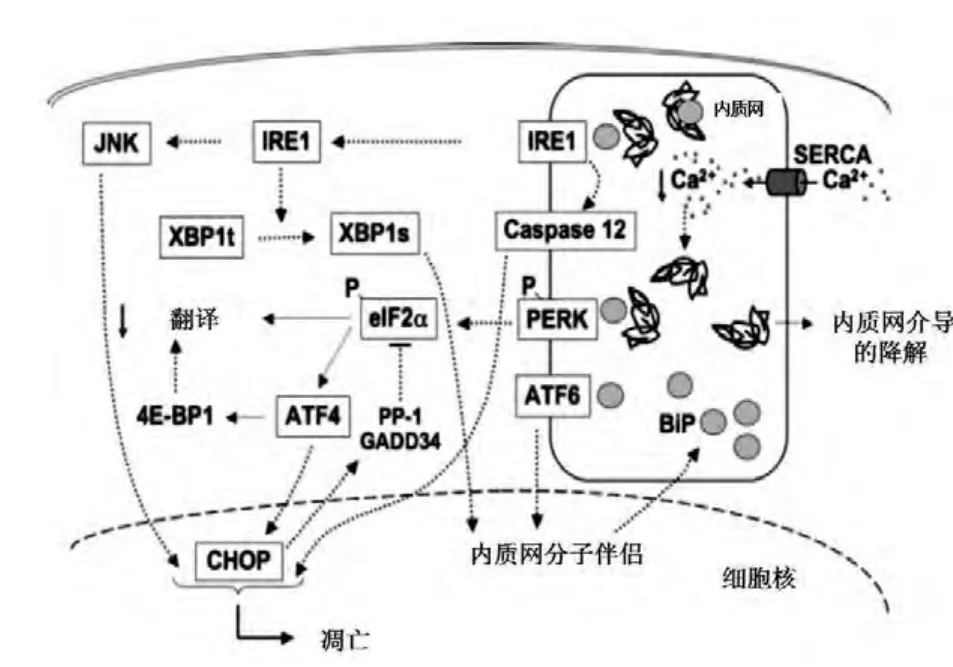
图1 ER应激信号转导途径[17]
胰岛β细胞大量合成胰岛素(占所有合成蛋白质的50%以上),并高水平表达ER应激的转换蛋白IRE1和PERK。临床中可以看到,PERK的基因(EIF2AK3)突变可导致新生儿及婴儿糖尿病(Wolscott-Rallison综合征)[18];编码ER Ca2+通道的WFS1基因突变导致Wolfram综合征伴糖尿病[19];而特异性针对β细胞的胰岛素基因突变也可导致新生儿糖尿病[20,21],这些突变可能是引起胰岛素的错误折叠从而引发ER应激,进而破坏胰岛细胞功能,导致β细胞凋亡和数量减少;动物研究显示,携带Ins2基因错义突变 (Cys96Tyr)的Akita小鼠自发出现高血糖和β细胞数量减少,而无胰腺炎和肥胖。在Akita小鼠发生糖尿病的过程中,ER分子伴侣BiP的mRNA表达增高,并诱导胰腺中ER应激相关凋亡因子CHOP的表达增加。体外试验中,小鼠MIN6 β细胞过表达突变的胰岛素insulin 2C96Y也诱导CHOP表达并导致β细胞凋亡;而破坏CHOP基因的表达可延缓杂合子Akita小鼠(Ins2WT/C96Y)糖尿病发病达8~10周[22]。
越来越多的研究表明,T2DM患者的胰岛β细胞存在ER应激,例如,T2DM患者的胰腺组织切片显示ER应激的标志物表达增高[23];T2DM患者β细胞的ATF3表达增高,eIF2α的下游蛋白ATF4[24]和CHOP[25]表达也增高。
研究发现[26],在INS-1细胞,棕榈酸和油酸诱导CHOP、ATF-4、BiP、XBP-1,激活ATF-6 启动子,提示这种FFA引发的ER应激反应可能在脂毒性所导致的β细胞凋亡的过程中发挥重要作用。研究显示[27,28],饱和脂肪酸所诱导的ER应激是重要的T2DM发生机制,棕榈酸诱导的β细胞凋亡主要是通过PERK依赖的CHOP途径介导的。
棕榈酸诱导的ER应激,分别经PERK-CHOP途径,通过抑制Bcl-2,诱导Caspase 3的表达而引发细胞凋亡;经IRE1途径,通过募集肿瘤坏死因子受体相关蛋白(TRAF2),激活下游JUN途径进而促进凋亡发生;此外,IRE1还可直接激活Caspase 12,引发β细胞凋亡[29]。
此外,ER应激还与高脂饮食喂养诱导的肥胖小鼠肝脏及脂肪组织的胰岛素抵抗有关[30];在肥胖者的脂肪和肝脏组织中也检测到ER应激标志物的表达增高[31,32]。因此,ER应激可能是T2DM两大病理生理紊乱,包括胰岛素抵抗和β细胞功能衰退的共同机制[33]。
利拉鲁肽保护胰岛β细胞功能
胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)受体激动剂是一类新的具有多种效应的抗糖尿病药物,这类药物通过刺激胰岛素分泌,抑制胰高糖素分泌等发挥降糖疗效。近年来人GLP-1类似物利拉鲁肽(Liraglutide)的相关研究显示,其减少β细胞凋亡的相关证据越来越多。
Friedreich共济失调(FRDA)是一种少见的由于Frataxin基因突变而致其功能丧失,从而导致呼吸链功能障碍而引发的一种神经变性疾病。该病可合并糖尿病(约占10%~30%),但机制不清。Cnop等[34]通过对41例FRDA患者的研究发现,患者的葡萄糖处置指数显著降低,提示存在胰岛β细胞功能失调;对部分病例的尸检胰腺病理切片分析,还发现FRDA患者胰岛中的β细胞数量减少。这一结论在Frataxin敲除的大鼠β细胞研究中也得到证实,Frataxin敲除的大鼠β细胞及人胰岛均有葡萄糖刺激的胰岛素分泌减少及β细胞凋亡,这可能与Frataxin缺乏使β细胞对油酸及ER应激诱导的凋亡更加敏感有关。而GLP-1和葡萄糖依赖性胰岛素释放多肽(GIP)可以保护保护Frataxin缺失的β细胞免于凋亡。
利拉鲁肽的体外研究[35],在高糖(25mmol/L)或正常糖(5mmol/L)条件下,分别加入利拉鲁肽或3-methyadenine分别孵育INS-1细胞24小时后检测细胞活力、自噬作用的相关指标,结果显示,高糖处理后INS-1细胞活力降低;自噬作用被抑制,INS-1细胞活力下降,凋亡也增加;而同时在高糖条件下加入利拉鲁肽INS-1细胞活力较单独高糖处理时有显著增加。因此认为利拉鲁肽可以保护INS-1细胞免受高糖诱导的凋亡以及与之相伴随的显著增加的自噬作用的影响。
用原代新生大鼠胰岛进行的体外研究显示[36],天然GLP-1和利拉鲁肽可以通过剂量依赖的方式抑制细胞因子和FFA诱导的胰岛β细胞凋亡;并且这一抗凋亡效应是通过GLP-1受体发挥作用的。在体的动物实验[37],在10周龄大的雄性db/db及m/m小鼠分别接受利拉鲁肽或生理盐水每日2次皮下注射共2周,比较治疗前后胰岛形态和生化、以及基因表达的改变情况。结果显示,利拉鲁肽改善db/db小鼠的代谢指标和胰岛素敏感性,其不仅增加葡萄糖刺激的胰岛素分泌(GSIS)及胰岛内胰岛素含量,降低胰岛内甘油三酯含量,还可下调促凋亡基因、ER应激及脂质合成的基因,上调细胞抗凋亡及抗氧化应激的基因等。形态学上的分析包括胰岛细胞的增殖、凋亡及氧化应激结果与基因分析结果相一致。
糖尿病前期的动物试验结果也显示[38],12周龄的OLEFT大鼠分别以利拉鲁肽50μg/kg、100μg/kg、200μg/kg剂量或生理盐水每日2次皮下注射共计12周,测定体重、摄食量、血脂、炎症标志物(纤维蛋白原、高敏C反应蛋白、IL-6、TNFα、PAI-1)、血糖、胰岛素敏感性及凋亡因子(Bcl-2和Bax)表达。结果发现12周龄的OLETF大鼠有显著的体重及摄食量增加,血脂水平升高,炎症因子及胰岛素水平增高。FPG水平显著增高但不超过7.0mmol/L且无IGT。治疗12周以后,对照组IFG、IGT、胰岛素抵抗、血脂、炎症状态均进一步恶化;胰岛体积增大且结构紊乱,炎性细胞浸润,而三组利拉鲁肽治疗组的IFG、IGT、增高的血脂及炎症标志物均得到逆转;胰岛素抵抗程度与治疗前水平相当;且利拉鲁肽恢复胰岛结构,上调Bcl-2表达,下调Bax表达,提示利拉鲁肽可能是通过调整凋亡因子及改善血脂代谢和炎症状态而恢复胰岛功能,从而抑制糖尿病前期的OLETF大鼠向糖尿病演变。
总结
在T2DM发生发展过程中,胰岛β细胞数量减少及β细胞功能紊乱是核心的致病因素,其中涉及各种不同的发生机制,其中近年来ER应激研究进展较快,并且ER应激被认为是β细胞功能衰退的重要原因之一。利拉鲁肽具有抗ER应激、促进β细胞增殖、分化,抗凋亡、抗氧化应激等多重作用从而抑制β细胞的凋亡,能够更好地保护胰岛功能和保存胰岛细胞数量,因此在T2DM治疗中具有美好的前景。
[1]Ferrannini E.Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes: problems and prospects.Endocrine Rev, 1998, 19(4): 477-490.
[2]Jallut D, Golay A, Munger R, et al.Impaired glucose tolerance and diabetes in obesity: a 6-year follow-up study of glucose metabolism.Metabolism, 1990,39(10):1068-1075.
[3]Defronzo RA.BantingLecture.From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus.Diabetes, 2009, 58(4):773-795.
[4]Spellman CW.Islet cell dysfunction in progression to diabetes mellitus.J Am Osteopath Assoc, 2007, 107(Suppl): S1-S5.
[5]Cnop M, Vidal J, Hull RL, et al.Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes.Diabetes Care, 2007, 30(3): 677-682.
[6]Cnop M, Igoillo-Esteve M, Hughes SJ, et al.Longevity of human islet α- and β-cells.Diabetes Obes Metab, 2011, 13(Suppl 1): 39-46.
[7]Cnop M, Hughes SJ, Igoillo-Esteve M, et al.The long lifespan and low turnover of human islet beta cells estimated by mathematical modeling of lipofuscin accumulation.Diabetologia, 2011, 53(2):321-330.
[8]Cnop M, Grupping A, Hoorens A,et al.Endocytosis of low-density lipoprotein by human pancreatic beta cells and uptake in lipid-storing vesicles, which increase with age.Am J Pathol, 2000, 156(1):237-244.
[9]Cnop M.Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes.Biochem Soc Trans, 2008, 36(Pt 3): 348-352.
[10]Kaiser N, Leibowitz G, Nesher R.Glucotoxicity and β-cell failure in type 2 diabetes mellitus.J Pediatr Endocrinol Metab, 2003, 16(1): 5-22.
[11]Cnop M, Welsh N, Jonas JC, et al.Mechanisms of Pancreatic β-Cell Death in Type 1 and Type 2 Diabetes:Many Differences, Few Similarities.Diabetes, 2005, 54(Supp 2): S97-107.
[12]Bachar E, Ariav Y, Ketzinel-Gilad M, et al.Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1.PLoS One, 2009, 4(3):e4954.
[13]Lenzen S.Oxidative stress: the vulnerable beta-cell.Biochem Soc Trans, 2008,36(Pt 3):343-347.
[14]Simmons RA.Developmental origins of diabetes: The role of oxidative stress.Best Pract Res Clin Endocrinol Metab, 2012, 26(5): 701-708.
[15]Pitocco D, Tesauro M, Alessandro R, et al.Oxidative stress in diabetes:implications for vascular and other complications.Int J Mol Sci, 2013, 14(11):21525-21550.
[16]Eizirik DL, Cardozo AK, Cnop M.The role for endoplasmic reticulum stress in diabetes mellitus.Endocr Rev, 2008, 29(1):42-61.
[17]Cnop M, Igoillo-Esteve M, Cunha DA, et al.An update on lipotoxic endoplasmic reticulum stress in pancreatic beta-cells.Biochem Soc Trans, 2008,36(Pt 5): 909-915.
[18]Delepine M, Nicolino M, Barrett T, et al.EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome.Nat Genet, 2000, 25(4):406-409.
[19]Inoue H, Tanizawa Y, Wasson J, et al.A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome).Nat Genet, 1998, 20(2):143-148.
[20]Colombo C, Porzio O, Liu M, et al.Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus.J Clin Invest,2008, 118(6):2148-2156.
[21]Stoy J, Edghill EL, Flanagan SE, et al.Insulin gene mutations as a cause of permanent neonatal diabetes.Proc Natl Acad Sci USA, 2007, 104(38): 15040- 15044.
[22]Oyadomari S, Koizumi A, Takeda K, et al.Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes.J Clin Invest, 2002,109(4): 525-532.
[23]Laybutt DR, Preston AM, Akerfeldt MC, et al.Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes.Diabetologia, 2007, 50(4):752-763.
[24]Hartman MG, Lu D, Kim ML, et al.Role for activating transcription factor 3 in stress-induced β-cell apoptosis.Mol Cell Biol, 2004, 24(13): 5721-5732.
[25]Huang CJ, Lin CY, Haataja L, et al.High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes.Diabetes, 2007, 56(8): 2016-2027.
[26]Kharroubi I, Ladriere L, Cardozo AK, et al.Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappa B and endoplasmic reticulum stress.Endocrinology, 2004, 145(11):5087-5096.
[27]Cunha DA, Hekerman P, Ladriere L, et al.Initiation and execution of lipotoxic ER stress in pancreatic β-cells.J Cell Sci, 2008, 121(Pt 14): 2308-2318.
[28]Pirot P, Ortis F, Cnop M, et al.Transcriptional regulation of the endoplasmic reticulum stress gene chop in pancreatic insulin-producing cells.Diabetes, 2007,56(4): 1069-1077.
[29]Cnop M, Ladriere L, Igoillo-Esteve M, et al.Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction.Diabetes Obes Metab, 2010,12(Suppl 2): 76-82.
[30]Ozcan U, Cao Q, Yilmaz E, et al.Endoplasmic reticulum stress links obesity,insulin action, and type 2 diabetes.Science, 2004, 306(5695): 457-461.
[31]Boden G, Duan X, Homko C, et al.Increase in endoplasmic reticulum stress related proteins and genes in adipose tissue of obese, insulin-resistant individuals.Diabetes, 2008, 57(9): 2438-2444.
[32]Sharma NK, Das SK, Mondal AK, et al.Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects.J Clin Endocrinol Metab,2008, 93(11): 4532-4541.
[33]Eizirik DL, Cardozo AK, Cnop M.The role for endoplasmic reticulum stress in diabetes mellitus.Endocr Rev, 2008, 29(1): 42-61.
[34]Cnop M, Igoillo-Esteve M, Rai M, et al.Central role and mechanisms of b-cell dysfunction and death in Friedreich ataxia–associated diabetes.Ann Neurol,2012, 72(6):971-982.
[35]Chen ZF, Li YB, Han JY, et al.Liraglutide prevents high glucose level induced insulinoma cells apoptosis by targeting autophagy.Chin Med J (Engl), 2013, 126(5):937-941.
[36]Bregenholt S, Moldrup A, Blume N, et al.The long-acting glucagon-like peptide-1 analogue, liraglutide, inhibits beta-cell apoptosis in vitro.Biochem Biophys Res Commun, 2005, 330(2):577-584.
[37]Shimoda M, Kanda Y, Hamamoto S, et al.The human glucagon-like peptide-1 analogue liraglutide preserves pancreatic beta cells via regulation of cell kinetics and suppression of oxidative and endoplasmic reticulum stress in a mouse model of diabetes.Diabetologia, 2011, 54(5):1098-1108.
[38]Guo N, Sun J, Chen H, et al.Liraglutide prevents diabetes progression in prediabetic OLETF rats.Endocr J, 2013, 60(1):15-28.
猜你喜欢
医学信息(2022年9期)2022-11-27
中国药学药品知识仓库(2022年1期)2022-03-23
中成药(2018年7期)2018-08-04
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
中成药(2016年8期)2016-05-17
中国卫生标准管理(2015年2期)2016-01-14
中国病理生理杂志(2015年8期)2015-12-21
中国药理学通报(2014年2期)2014-05-09
中国医学科学院学报(2014年6期)2014-03-11