
有机磷酸稳定剂对双氧水绝热分解特性的影响
2014-07-24 10:29朱希增朱顺兵刘新华
化工进展 2014年12期
朱希增,朱顺兵,刘新华
(南京工业大学城市建设与安全工程学院,江苏省危险化学品本质安全与控制技术重点实验室,江苏 南京210009)
工业级的H2O2中常常由于金属杂质以及温度变化等因素,促使双氧水发生分解放热,给生产、使用和储存带来不利影响,其中金属杂质以Fe3+最为常见,影响也更为严重,因此必须加入稳定剂来抑制其催化分解作用。有机磷酸稳定剂对Fe3+、Ca2+、Mg2+等离子具有优异的螯合能力,能与金属离子螯合形成稳定的水溶性螯合物,同时本身也极为稳定,在纺织工业、冶金和化学分析上都有重要的应用[1-3]。目前,国内外学者[4-6]做了大量的双氧水与有机溶剂、酸碱、金属杂质的相容性研究,但稳定剂与双氧水作用的实验文献极少。孙峰等[6]研究了不同质量分数Fe3+对双氧水绝热分解的影响,得到工业储存条件下Fe3+质量分数的安全临界值,但是并未对双氧水稳定剂进行研究。1979年,华东化工学院与上海桃浦化工厂[7]开展了将有机多元磷酸(ATMP 等)作为过氧化氢稳定剂的试验研究。采用简单方法研究了不同浓度的有机磷酸抑制温度、金属离子对H2O2的分解。但是,这种方法仅仅得到了过氧化氢的分解率变化,并未揭示实际情况下的热危险性的表征。针对此现象,本文研究在绝热条件下有机磷酸稳定剂对双氧水稳定性的影响,旨在为双氧水的安全生产、运输及储存提供 参考。
1 实验部分
1.1 实验样品
质量分数为30%的双氧水,分析纯,AR,国药集团化学试剂有限公司。FeCl3·6H2O,分析纯,AR,含量≥99%,国药集团化学试剂有限公司。质量分数为50%的氨基三亚甲基磷酸(ATMP),阿拉丁试剂,用去离子水稀释至所需浓度供测试使用。
1.2 实验仪器
VSP2 是由美国Fauske&Associates(FAI)公司生产,可用于获得热动力学和热危害表征参数[8-10]。样品测试池分为闭口和开口两种,可用于不同类型的量热实验。测试池位于反应釜内部,两者之间用隔热棉填充。由于测试池的热惰性很低(接近1),可直接放大至工业尺寸,无须经过繁琐的修正计算。
VSP2 采用“H-W-S”(加热-等待-搜索)模式,样品首先在绝热条件下被预加热到设定的温度值,稳定一段时间(一般为5min)以消除热惯性,达到热平衡后,通过对比升温速率和温度灵敏度判断是否有放热。如果VSP2 未检测到样品放热,则通过加热器使反应系统温度升高一个台阶(5℃),进入下一个“加热-等待-搜索”循环,直到检测到样品的放热现象。表1 列出了VSP2 试验条件。当系统探测样品开始放热时停止加热,样品在绝热环境下靠自分解放热升温。同时系统自动记录下时间、温度、压力、绝热温升速率、压升速率等数据。

表1 VSP2 试验条件
1.3 实验方法
为了防止测试池爆裂或严重变形导致实验获得的数据不准确,VSP2 采用低浓度或较少质量的样品来进行测试。本实验对含15%双氧水的样品开展了封闭系统量热实验,并在恒定背压(3MPa)下,对含27.5%的双氧水进行开放系统量热测试。表2列出了实验样品的组成及质量分数。
掺杂了Fe3+的双氧水极易发生分解,样品很可能在测试程序开始之前就已经反应,导致测试的T0偏高。因此试剂均被储存于冰箱中,维持恒温10℃。为保证测试结果的准确性,样品导入测试池后,先不开主加热器进行加热,仅打开辅助加热器维持反应体系处于绝热环境中,在该温度下稳定10min。如果检测到放热,关闭主加热器和“H-W-S”模式,切换到绝热追踪模式。反之按预先设置的“H-W-S”模式进行测试。
实验所用测试池的质量为39g,比热容约为0.427J/(g·K),双氧水比热容为4J/(g·K)。
热惰性因子计算如式(1)[11]。

可以得到热惰性因子Ф=1.1。
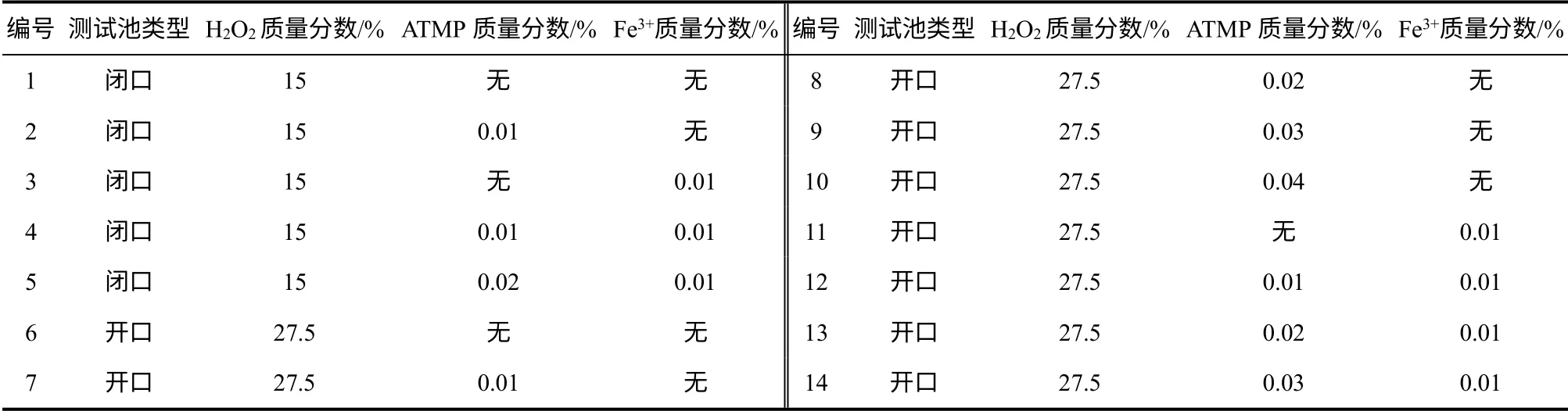
表2 试验样品组成
2 结果与讨论
2.1 绝热分解特性参数
2.1.1 有机磷酸对不同温度影响双氧水分解的抑制作用
温度是影响双氧水分解的重要因素。根据范特霍夫定律,温度每升高10K,分解速度将增加2~4 倍。初始放热温度是物质发生热反应时,衡量该物质发生反应难易程度的重要参数。图1 为加入稳定剂前后双氧水绝热分解过程的温度-时间及压力-时间曲线。表3 为加入稳定剂前后双氧水绝热分解参数的数据。
如图1(a)中的温度-时间曲线所示,15%H2O2在50℃时发生分解,在其他实验条件均不变的情况下加入0.01%ATMP,其T0升高为80℃。因为有机磷酸稳定剂具有吸附和去活化作用,使反应活化能升高,导致T0提高,其稳定性得到增强。
从表3 可以看出,15%双氧水的Tmax、(dT/dt)max、(dp/dt)max分别为140℃、52℃/min、3.95MPa/min。在ATMP 的作用下,其值分别增加至169℃、72℃/min、6.22MPa/min。

图1 加入稳定剂前后双氧水绝热分解过程的温度-时间及压力-时间曲线

表3 加入稳定剂前后双氧水绝热分解参数
这是因为双氧水在低温下只发生非常缓慢的催化分解反应,此时放热速率低于VSP2 的温度灵敏度,无法检测到放热,但该过程中双氧水的浓度逐渐降低,从而导致样品随后的反应时间延长,温升速率、压升速率降低。另外,由于实验设备无法做到绝对的绝热,在反应过程中有热量的损失,不含稳定剂的双氧水分解反应过程时间长,体系传递到环境的热量多,得到的最高温度Tmax较低。
由图2 可知,样品的起始分解温度随ATMP 质量分数的增加而增大,在相同ATMP 增长量的基础上,T0的增长幅度逐渐减缓。当双氧水中ATMP 质量分数从0 升高为0.04%时,其初始分解温度T0由50℃提高至115℃,稳定剂浓度不同对双氧水稳定性的影响不同,T0达到115℃。
2.1.2 有机磷酸浓度对Fe3+催化双氧水分解的抑制作用
从图3 和表4 可以看出,当Fe3+与ATMP 浓度比为1∶1 时,最大温升速率和最大压升速率分别为37℃/min、2.5MPa/min。当Fe3+与ATMP 浓度比为1∶2 时,最大温升速率和最大压升速率分别升高为40℃/min、2.9MPa/min。
这是因为当Fe3+加入到H2O2后,双氧水在低温下只发生非常缓慢的催化分解反应,此时放热速率很低,VSP2 无法检测到细微的温度变化,又因为在该过程中双氧水的质量分数逐渐降低,导致样品随后的反应速率降低、反应时间延长、自分解放热速率降低。
由图4 可知,随着ATMP 质量分数的增加,样品的T0随之升高,但T0增长幅度逐渐减缓。当ATMP质量分数为0.03%时,T0几乎不再上升。

图2 不同质量分数ATMP 对双氧水的T0 影响

图3 不同质量分数ATMP 与含Fe3+的双氧水作用的温度-时间及压力-时间曲线

表4 不同质量分数ATMP 与含Fe3+的双氧水作用的绝热分解表征参数
敞开系统量热测得的T0与封闭系统量热相比,其值偏高。这是因为含铁离子的双氧水在温度较低的时候存在缓慢的分解,放出微量的热量,之后又被气体通过开口带出测试池,无法被VSP2 侦测,导致温度监控系统检测到反应的放热延后。无铁离子的样品起始分解温度高,气体带走的热量对放热的检测无影响。

图4 不同质量分数ATMP 对含Fe3+的双氧水的T0 影响
由图2、图4 可以看出,在ATMP 作用下,双氧水以及掺杂了Fe3+的双氧水的起始分解温度均有所提高。分别在质量分数为0.01%、0.02%、0.03%ATMP 的影响下,双氧水的起始分解温度均高于掺杂了Fe3+的双氧水的起始分解温度,说明有机磷酸稳定剂不能完全消除Fe3+对双氧水的分解作用。
2.2 动力学分析
绝热过程的反应动力学参数是评价物质危险性的重要依据,根据阿伦尼乌斯公式,见式(2)。

式(2)两边取对数,有式(3)。

假设加入Fe3+及稳定剂的双氧水分解为一级反应,取n=1,则有式(4)。

将VSP2 测试所得到的绝热分解参数代入式(4),可以得到k 随T 的变化关系[12],从图5 可以看出,lnk 与-1000/T 关系近似为直线,验证假设成立,反应均为一级反应。根据式(3),通过其直线的截距和斜率求出A 和Ea。反应的A 和Ea计算结果如表5 所示。
从表5 可以看出,15%H2O2的活化能为92 kJ/mol,指前因子为4.17×1010;含Fe3+的质量分数为0.01%时的双氧水活化能为72.8kJ/mol,指前因子为5.12×108;随着ATMP 质量分数的增加,活化能与指前因子均有提高。表明在有机多元磷酸稳定剂作用下,双氧水以及在掺杂Fe3+的情况下的表观活化能升高,分解反应越难进行,要求反应温度越高,发生热失控的风险越低。

图5 lnk 与-1000/T 的关系

表5 样品反应动力学参数
2.3 绝热条件下热爆炸的形成时间
反应热风险是由反应失控及其相关后果带来的风险,可以从两方面来评估:严重度和可能性。化工工艺过程中最危险的情况是反应器冷却失效,即反应物料处于绝热状态。因此,经常利用绝热温升来作为评估严重度的判据。另外,化工风险的可能性可通过T0和TMRad作为时间尺度进行评估[13]。
绝热条件下到达最大反应速率时间(TMRad)是判断物质发生热失控可能的重要依据,在工业上常常被当作最危险情况下的紧急响应时间,可用于表示采取措施避免事故发生或最大限度降低事故造成伤害的时间[15],是热灾害评估中的一个非常重要的参数。失控可能性评估标准如表6 所示。
计算公式如式(5)[16]。

由于样品的含能物质相同,反应过程中的绝热温升ΔTad均在90℃左右,因此得式(6)。

从表7 可以看出,当存储温度为25℃时,15%的双氧水的TMRad为7.1h,失控可能性为“很可能”,稳定性不高;当双氧水中ATMP 质量分数为0.01%时,到达最大反应速率时间有明显推迟;当双氧水中掺杂0.01%Fe3+时,TMRad仅为0.32h,留给操作人员的反应时间极短,一旦反应发生失控,则极易造成失控反应事故的发生。随着ATMP 加入量的增加,TMRad也随之升高。因此对于无论是否掺杂Fe3+的双氧水,稳定剂ATMP 都可以提高双氧水的存储稳定度,并且随其在双氧水中质量分数的增加,发生失控的可能性降低。

表6 反应失控可能性评估标准[14]

表7 样品失控可能性评估(T0=25℃)
3 结 论
(1)双氧水的初始分解温度为50℃,活化能为92kJ/mol,指前因子为4.17×1010,在25℃下到达最大温升速率时间为7.1h。在有机磷酸稳定剂的影响下,双氧水的T0升高到80℃,并随着ATMP 质量分数的增加而提高,但增加幅度逐渐减缓。同时,活化能和指前因子分别提高到111.7kJ/mol 和4.03×1014,TMRad达到67.8h。稳定剂对温度变化使双氧水分解的抑制作用明显。
(2)掺杂了0.01%Fe3+的双氧水在常温下就发生分解反应,活化能为72.8kJ/mol,指前因子为5.12×108,在初始温度为25℃时的TMRad为0.32h。与0.01%Fe3+的H2O2相比,H2O2与ATMP 混合后,起始分解温度有明显提高。随ATMP 质量分数的增加,其活化能与指前因子均有大幅提高,失控可能性降低为“偶尔”。稳定剂能有效延缓Fe3+对双氧水的催化分解作用。
(3)针对双氧水的起始分解温度,ATMP 存在一个作用上限质量分数。此时继续增加ATMP 的质量分数,双氧水的起始分解温度几乎就不再上升。
(4)有机磷酸稳定剂使双氧水的T0、TMRad有不同程度的提高,并且各组样品测试得到的ΔTad均在90℃左右。因此,在潜在的严重性不变的前提下,事故发生的可能性降低,表明有机多元磷酸稳定剂能使双氧水发生失控的热风险降低。
符 号 说 明
A——指前因子,s-1
C0n-1——反应物的初始浓度,mol·L-1
cp——比热容,J·g-1·℃-1
cvb——测试池的平均比热容,J·g-1·K-1
cvs——反应物的平均比热容,J·g-1·K-1
Ea——活化能,kJ·mol-1
k——反应速率常数,s-1
mb——测试池质量,g
ms——反应物的初始质量,g
n——反应级数,量纲为1
pmax——最高压力,MPa
Q——反应热,J·g-1
R——气体常数,8.314J·mol-1·K-1
T——温度,℃
TMRad——到达最大反应速率时间,h
ΔTad——绝热温升,℃
Tf——失控反应的最终温度,℃
Tmax——最高温度,℃
T0——起始放热温度,℃
Ф——热惰性因子,量纲为1下角标
ad——绝热状态
b——测试池
f——最终的
s——反应物
max——最大值
[1] 赵惠敏. 双氧水稳定剂的研究进展[J]. 化工科技,2003(2):55-59.
[2] 李文斌,张运刚,袁华. 过氧化氢复配稳定剂研究[C]//中国化学会第五届全国化学推进剂学术会议,2011.
[3] 徐沛楷. 新型多用途的螯合剂——有机膦酸类化合物[J]. 化学试剂,1981(6):25-30.
[4] Mackenzie J. Hydrogen peroxide without accidents[J]. Chemical Engineering,1990,97(6):84-90.
[5] Papadaki M,Gao J,Mahmud T,et al. Catalytic decomposition of hydrogen peroxide in the presence of alkylpyridines : Runaway scenarios studies[J]. Journal of Loss Prevention in the Process Industries,2005,18(4-6):384-391.
[6] 孙峰,谢传欣,黄飞,等.Fe3+掺杂对双氧水热稳定性的影响[J]. 安全与环境学报,2010(4):176-180.
[7] 汪祖模,徐玉佩,徐国强,等. 有机多元膦酸用作工业过氧化氢稳定剂的研究[J]. 化学世界,1981(3):11-12.
[8] 刘显凡,化学反应放热失控安全泄放设计及评估技术研究[D]. 大连:大连理工大学,2011.
[9] Shu C M , Yang Y J. Using VSP2 to separate catalytic and self-decomposition reactions for hydrogen peroxide in the presence of hydrochloric acid[J].Thermochimica Acta,2002,392:259-269.
[10] 丁凌云,周贤太,纪红兵. 有机过氧化物热动力学及热危险性研究进展[J]. 化工进展,2011,30(11):2369-2375.
[11] Leung J C,Fauske H K,Fisher H G.Thermal runaway reactions in a low thermal inertia apparatus[J]. Thermochimica Acta,1986,104:13-29.
[12] Chi J H,Wu S H,Shu C M,et al. Thermal hazard accident investigation of hydrogen peroxide mixing with propanone employing calorimetric approaches[J]. Journal of Loss Prevention in the Process Industries,2012,25(1):142-147.
[13] 程春生,秦福涛,魏振云. 化工安全生产与反应风险评估[M]. 北京:化学工业出版社,2011.
[14] Stoessel F. What is your thermal risk?[J]. Chemical Engineering Progress,1993,89(10):68-75.
[15] Wu S H,Chi J H,Huang C C,et al.Thermal hazard analyses and incompatible reaction evaluation of hydrogen peroxide by DSC[J].Journal of Thermal Analysis and Calorimetry,2010,102(2):563-568.
[16] Stoessel,F.Thermal Safety of Chemical Processes:Risk Assessment and Process Design[M].Weinheim,Germany:Wiley-VCH,2008.
猜你喜欢
中国新闻周刊(2022年16期)2022-05-09
石油沥青(2021年4期)2021-10-14
趣味(语文)(2020年6期)2020-11-16
广州化工(2020年5期)2020-04-01
中国资源综合利用(2016年6期)2016-01-22
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
中国塑料(2015年3期)2015-11-27
中国塑料(2015年3期)2015-11-27
中国储运(2015年3期)2015-11-22
中国塑料(2015年10期)2015-10-14
