
采用高效液相一测多评法对保和丸中陈皮和连翘的含量测定研究
2014-07-21 09:12:32张秀丽
山西卫生健康职业学院学报 2014年3期
杨 艳,张秀丽
(1.山西省食品药品检验所,山西太原 030001;2.繁峙县人民医院,山西繁峙 034300)
保和丸始载于《丹溪心法》,最早为水丸,由焦山楂、炒六神曲等8味药组成,具消食、导滞、和胃作用,主要用于食积停滞、脘腹胀满等症。现有161个生产企业,216个批准文号,大、小蜜丸、水丸和浓缩丸4种剂型。浓缩丸执行标准为《卫生部药品标准》中药成方制剂第七册,除性状外,仅收载有3味药的显微鉴别。为控制本产品质量,故拟增加非标方法陈皮中橙皮苷及连翘中连翘酯苷A的含量测定项。
1 仪器与试药
仪器:WATERS Acquity UPLC超高效液相色谱仪,二极管阵列检测器(沃特世科技有限公司)。
试药:乙腈为色谱纯;水为纯化水;冰醋酸为分析纯;其他试剂均为分析纯。
对照品:连翘酯苷A、橙皮苷(供含测用,批号分别为L-012-110921、110721-200613),均购于中国药品生物制品检定所。
样品:药都制药集团股份有限公司(生产批号:110304)。
2 方法与结果
2.1 色谱条件
2.1.1 色谱柱的选择 a)资生堂 C18柱(5 μm,4.6 mm ×250 mm);b)自装柱富士填料 C18(5 μm,4.6 mm × 250 mm);c)Allitima C18(5 μm,4.6 mm ×250 mm)。分离良好,柱温: 室温,流 速:0.7 mL·min-1。
2.1.2 流动相的选择 实验过程中共选择了以下2种流动相系统:
a)流动相A为乙腈,流动相B为0.5%冰醋酸溶液,线性梯度洗脱:0 min— A∶B=10∶90;60 min—A∶B=50∶50;b)流动相A为甲醇,流动相 B为0.5%冰醋酸溶液[1],线性梯度洗脱:0 min—A∶B=10∶90;60 min— A∶B=50∶50。
结果表明,流动相系统中以第一种,即流动相A为乙腈,流动相 B为0.5%冰醋酸溶液,梯度洗脱0 min—A∶B=10∶90;60 min— A∶B=50∶50为最佳,谱图上各个色谱峰的分离度较好,保留时间适中。因此选此流动相作为保和丸的含量测定的流动相(见表1)。
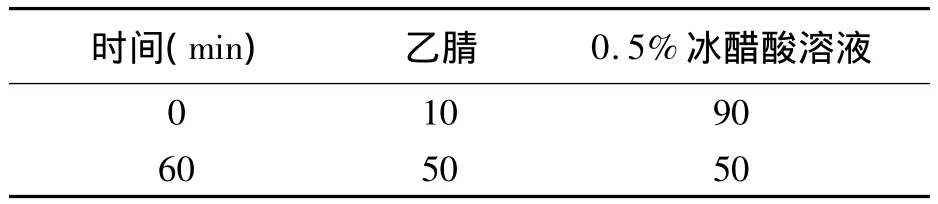
表1 流动相 %Table 1 Mobile Phase %
结果:采用以上色谱柱及流动相均能达到良好的分离效果(见图1~3)。

图1 连翘酯苷A对照品HPLC色谱图Fig.1 HPLC Chromatogram Map of Forsythiaside A Controls

图2 橙皮苷对照品HPLC色谱图Fig.2 HPLC Chromatogram Map of Hesperidin Controls

图3 保和丸供试品HPLC色谱图(1.连翘酯苷A对照品,2.橙皮苷对照品)Fig.3 Baohe Pills and Samples HPLC Chromatogram Map(1.horsythiaside A controls,2.hesperidin controls)
2.1.3 检测波长的选择 参照《中国药典》2010年版一部连翘药材、陈皮药材[含量测定]项下方法[2],选择二极管阵列检测器,波长范围190 nm~400 nm,提取280 nm波长处的色谱图,色谱图分离良好。
2.2 供试品提取方法的考察
2.2.1 提取溶剂的考察 分别采用50%甲醇、70%甲醇、85%甲醇、正丁醇、乙酸乙酯、甲醇超声提取及加热回流法[3],通过比较,采用70%甲醇加热回流提取法效果最佳。
2.2.2 提取时间的考察 采用加热回流提取法,分别加热30 min、45 min、60 min,结果见表2。
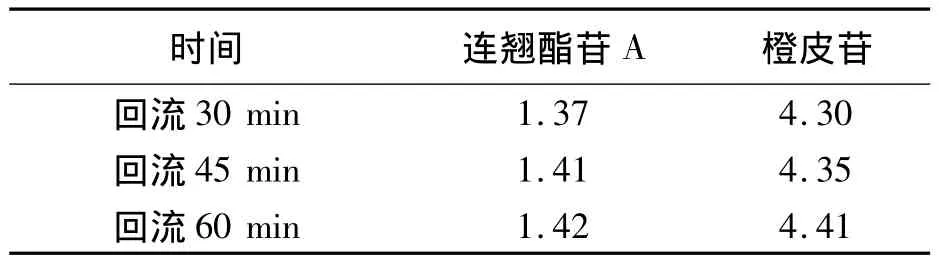
表2 提取时间考察结果 mg·g-1Table 2 The Inspection Results of Extraction Time mg·g-1
结果表明:加热回流45 min与60 min含测结果虽相差不多,但考虑到能使大蜜丸充分溶散,故将加热回流时间定为60 min。
2.3 专属性实验
分别取处方中除连翘、陈皮外的全部药味,按本品工艺及供试品溶液的制备方法制备阴性对照溶液,注入液相色谱仪进行测定,结果表明:在连翘酯苷A、橙皮苷保留时间处无干扰。
2.4 耐用性实验
用不同仪器:日本岛津LC-2010AHT型高效液相色谱仪,Agilent1200高效液相色谱仪;不同色谱柱:a)资生堂 C18柱(5 μm,4.6 mm×250 mm);b)自装柱 富 士 填 料 C18(5 μm,4.6 mm× 250 mm);c)AllitimaC18(5 μm,4.6 mm ×250 mm),均能达到良好的分离效果。
2.5 线性关系的考察
精密称取连翘酯苷A对照品9.56 mg,置100 mL量瓶中,加70%甲醇使溶解并稀释至刻度,摇匀,即得。另精密称取橙皮苷10.18 mg,置50 mL量瓶中,加70%甲醇使溶解并稀释至刻度,摇匀,即得。按不同进样量依法测定,以峰面积值为纵坐标,进样量为横坐标,绘制标准曲线,测得连翘酯苷A回归方程为:A=1E+0.6X+806.4,r2=0.999,表明在0.019 ~ 0.956 μg范围内有良好的线性关系;橙皮苷回归方程为:A=3E+0.6X+32 918,r2=0.999,表明在0.102 ~ 2.036 μg范围内有良好的线性关系。
2.6 精密度考察
取同一供试品(批号110304)溶液,同一进样量,连续进样6次,按实验条件依法测定,结果RSD连翘酯苷A为2.2%,橙皮苷为0.3%,符合要求。
2.7 稳定性考察
取同一供试品(批号110304)溶液,每隔一定时间同一进样量,按实验条件依法测定,结果表明供试液在36 h内稳定,RSD分别为:连翘酯苷A为2.7%,橙皮苷为1.0%,符合要求。
2.8 重复性考察
按拟订含量测定方法,对同一批号(批号110304)的6份样品进行平行实验,依法测定,平均含量连翘酯苷 A 为0.684 5 mg·g-1,橙皮苷为4.358 0 mg·g-1;RSD:连翘酯苷A为1.4%,橙皮苷为0.79%。
2.9 准确度实验
采用加样回收法,取上述重复性实验所用的样品(1103041批),取1 g(9份),精密称定,分别精密加入连翘酯苷 A 对照品约0.5 mg、0.7 mg、0.8 mg,橙皮苷对照品约3.4 mg、4.3 mg、5.3 mg,按拟定含测方法制备,依法测定,计算回收率,结果见表3。
回收率=[测得总量-(取样量×重复性测得含量)]/加入对照品量×100%。
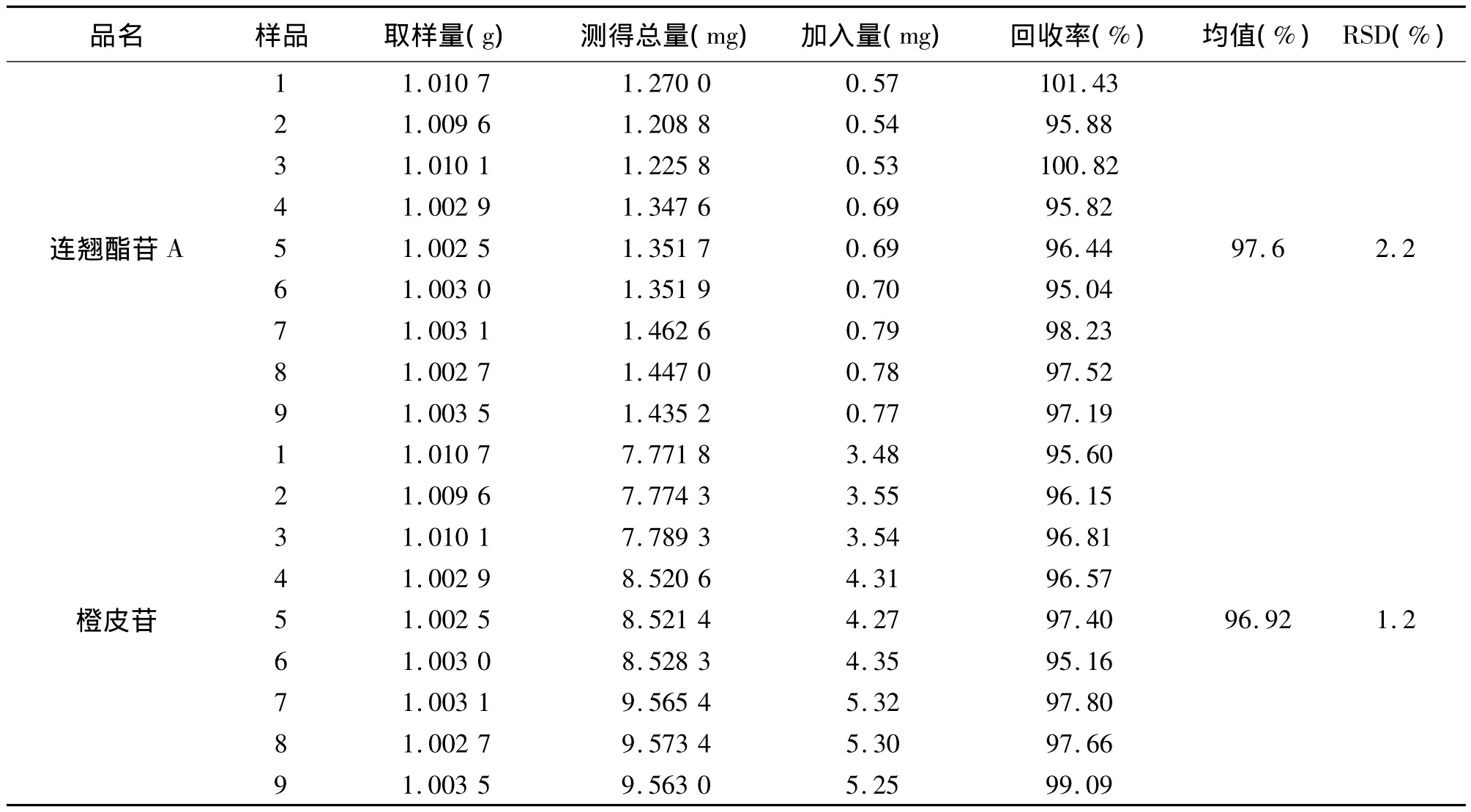
表3 回收率测定结果Table 3 Determination Results of Recovery Rate
2.10 限度确定
根据中国药典2010年版一部项下规定,结合连翘、陈皮含量限度,按处方量及转移率30%计算,暂定限度为:本品每克含连翘以连翘酯苷A计,不得少于0.1 mg·g-1,含陈皮以橙皮苷(C28H34O15)计,不得少于1.2 mg·g-1。
3 样品的测定
依法对107批次样品进行测定,测得结果分别为橙皮苷:最低0.232 mg·g-1,最高4.871 mg·g-1,107批次浓缩丸50批次不合格,占46.7%;连翘酯苷A最低0.064 mg·g-1,最高2.391 mg·g-1,107 批次浓缩丸6批次不合格,占5.6%。
4 讨论
4.1 标准项目缺失比较严重
不仅无法实现本品的标准可控和质量安全有效的目标,而且难以发现以次充好、以假充真、偷工减料等违法行为。因此,必须进一步提高和完善本品的现行标准。
4.2 部分药材质量状况堪忧
本次抽验107批次样品标准检验和探索研究发现,虽未发现擅自不按处方投料、改变工艺等违法行为,但质量方面的问题比较突出。
4.3 先进技术手段明显缺乏
本品现行标准仅有显微鉴别,未能采用一测多评等专属性强、灵敏度高的先进技术手段和方法进行鉴别和含量测定,无法实现对本品整体质量的有效控制。因此,本品现行标准中应积极引进先进的技术手段和方法,以确保质量控制的全面准确和科学深入。
[1]梁 艳,郜凤香.高效液相色谱法测定木香分气丸中橙皮苷含量[J].中国药业,2013,22(8):52-53.
[2]国家药典委员会.中华人民共和国药典(2010年版一部)[S].北京:中国医药科技出版社,2010.
[3]孟 和,朱艳红,吴学军.HPLC法测定保和丸中连翘苷的含量[J].中国药事,2013,27(3):312-314.
猜你喜欢
人人健康(2021年14期)2021-08-06 08:58:16
中国民间疗法(2021年8期)2021-07-22 05:53:08
时代邮刊(2019年22期)2019-12-17 14:40:36
时代邮刊·下半月(2019年11期)2019-09-22 06:09:21
陕西中医(2018年6期)2018-08-29 00:43:24
益寿宝典(2017年2期)2017-02-26 21:27:52
吉林大学学报(医学版)(2015年3期)2015-12-17 07:47:51
健康必读(2015年4期)2015-06-01 02:11:05
中国当代医药(2015年24期)2015-03-01 02:06:02
中国当代医药(2015年9期)2015-03-01 02:02:08
