
Genomic Islands Prediction and Analysis in Cyanobacteira by Bioinfomatics
2014-07-14 01:20YiLiNiNiRaoFengYangandHanMingLiu
Yi Li, Ni-Ni Rao, Feng Yang, and Han-Ming Liu
1. Introduction
Cyanobacteria are a diverse group of photoautotrophic bacteria, which have multiple phenotypic characteristics and typical distribution pattern. Their remarkable developed adaptations enable them to colonize variable and extreme habitats in earth ecosystems and cause their divergent sequences[1]. Prochlorococcus is one of the representative genera that exist in the oceans, contributing significantly to primary metabolism production. Two different light-adapted ecotypes (high-light and low-light),span most of the surface oceans and differ from each other in their geographic distribution. The genomes belonging to the same species or ecotypes are closely related, which generally have large syntenic regions, and the conservative core genes and their members are arranged as a fixed order in these sections. However, these areas are frequently interrupted by genomic islands. Genomic islands (Gis) are defined as horizontally acquired genomic regions, and their mutations usually interrupt or destroy the original mode of gene expression and signal transformation[2]. Gis are often greater than 8 kB and non-conservative genes involved distribute randomly. Besides, Gis also contain many horizontal transfer genes and insert fragments, which increase intraspecies variability.
Gis are important for genomic modulation of bacteria.They are elements of the flexible gene pool and have a wide range of functions, such as iron uptake[3], toxin production[4], and nitrogen fixation[5]. Gis are demonstrated to play a key role as a repository for transferred genes,genome size modulation, and stress adaption in marine Synechococcus[6]. When whole-genome alignment is carried out in Prochlorococcus marinus MED4 and Prochlorococcus marinus str. MIT 9312, five islands are discovered[7]. Three of them are confirmed to be hypervariable while the other two are relatively stable.Different island genes accompany with distinct functions.For example, the genes in ISL3 act on photo inhibition and nutrient assimilation, but those in ISL5 still have functions related to adaptation of phosphorus starvation. Thus, we can quickly locate the rough position of a new ncRNA if we know its function.
In this study, we combine algorithms, software, and sequence features to predict the genomic islands of Synechocystis sp. PCC 6803, which is a cyanobacterial strain. Besides, we observed that the hypervariable positions of genomic islands are out of proportion to their homology, and different sRNAs are mapped in different strains. Thus, we pick two typical sRNA, Yfr1 and Yfr7, to explain the reason.
2. Materials and Methods
Sequenced genomes are downloaded from NCBI,Synechocystis sp. PCC 6803, GI: NC_000911;Prochlorococcus marinus MED4, GI: NC_005072;Prochlorococcus marinus SS120, GI: NC_005042;Prochlorococcus marinus str. AS9601, GI: NC_008816;Prochlorococcus marinus str. MIT 9515, GI: NC_008817.First, Gis are predicted by using SIGI-HMM[8], IslandPath-DIMOB[9], and IslandPick[10], respectively, and the results are merged. Then, we screen them by some sequence features like composition bias, adjacent to tRNA, integrases,transposases, inserted sequence (IS), and direct repeat (DR)to pick out the most possible Gis.
Mauve[11]is used to do whole genome alignments and view homologous regions, then gene rearrangement and horizontal transfer are analyzed combined with HGT-DB(horizontal gene transfer database).
3. Results
SIGI-HMM, IslandPath-DIMOB, and IslandPick are implemented to predict Gis based on hidden Markov models, GI-associated features and comparative genomics approach respectively. The former two kinds of software are shown to have a higher specificity and accuracy in prediction[10]. However, IslandPick is included since it is not based on sequence composition and shows more agreement with distantly related strains. We used them to predict Gis in Syn. PCC 6803 and merged the results, as shown in Table 1. We can see that only five Gis are longer than 10 kb. The longest one reaches 37916 bp with 46 protein-coding genes while the shortest one only has 4812 bp with 3 genes, which indicates Gis have a wide range of length and constitution. Gis can be classified as pathogenicity islands, resistance islands, xenobioticdegradation islands, metabolic islands, symbiosis islands,and fitness islands according to their diverse functions. We analyze the results and find that most of types are included in Syn. PCC 6803.
Pathogenicity islands are known as their virulence factors which mainly include adhesin, invasin, endotoxin,and exotoxin, and they are found in bacteria, plant, and animal. Due to the properties of pathogenicity islands defined by Ohad Gal-Mor[12], we did not found virulence factors on Gis of Syn. PCC 6803. Although two virulence-associated proteins (sll0690 and ssl2922) exist in genome, they are discrete and parts of toxin/antitoxin operon, and their concentration is low under normal condition, which is not enough to be a factor of pathogenicity islands. Thus, we believe that Syn. PCC 6803 dose not have pathogenicity islands while they can produce toxins in some special conditions.
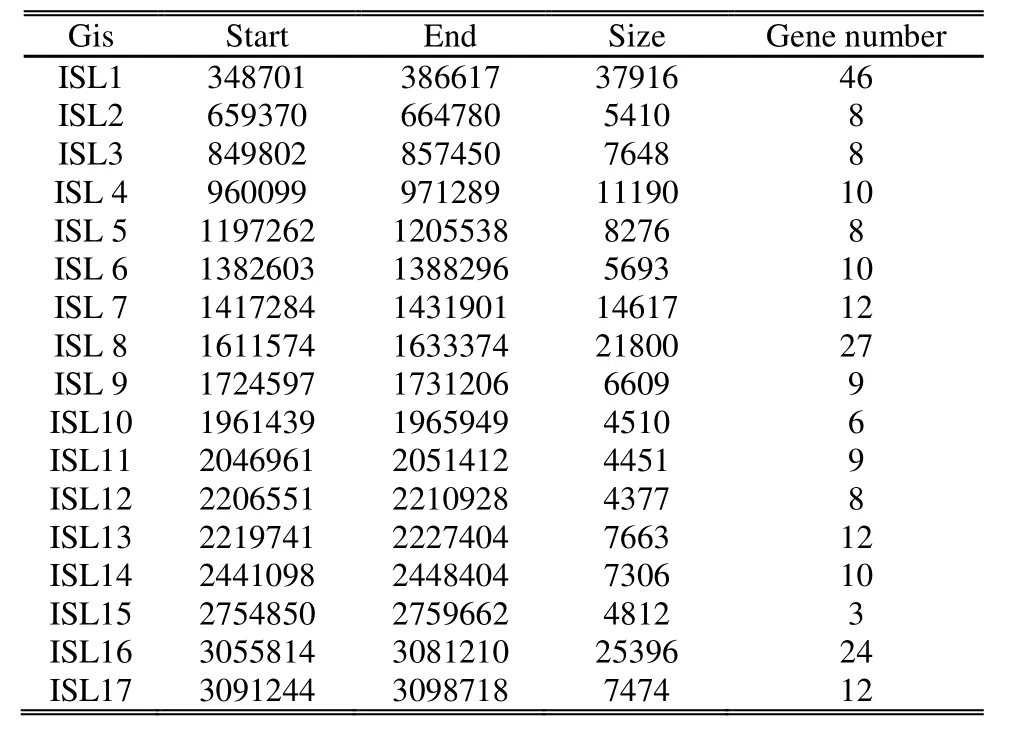
Table 1: Genomic islands predicted in Syn. PCC 6803
Resistance islands usually participate in antibiotic resistance with multiple resistance loci. They exist in plasmid, integron, super-integron, and composite transposon at first, and then discontinuous segments are reintegrated by these movable components and genes are gathered together to transfer as an entirety, which form the resistance islands. In our results, the quinolene resistance gene (norA), nickel resistance gene (nreB), and mercuric resistance gene (merR) are found in ISL6, ISL16 and ISL17,respectively. NorA produces resistance to norfloxacin and other hydrophilic quinolones and reduces norfloxacin accumulation in intact cells that are energy dependent,suggesting active drug efflux as the mechanism of resistance. Nickel binds to proteins and nucleic acids and frequently inhibits enzymatic activity, DNA replication,transcription, and translation. However, the excess nickel is toxic and its concentration can be balanced by nreB. The merR protein mediates the induction of the mercury resistance phenotype and plays a central role in the heavy metal responsive system of bacteria. These resistance islands are becoming more important with the increasing seriousness of water pollution.
The first metabolic island was known for the conjugative transposon CTnscr94 discovered in Salmonella senftenberg by Hochhut[13]. CTnscr94 has been proved to transfer to Escherichia coli K-12 independently and 3’end of tRNA-Phe is one of the integration sites. In our results,ISL8 and ISL11 contain tRNA-Phe and tRNA-His, and most of these genes play a role in the regulation of cell metabolism. For instance, slr2107 and slr2108 on ISL8 participate in the transportation of lipopolysaccharide, and slr0611 on ISL11 participates in the biosynthesis of terpenoid backbone. We note that at least one gene in each Gi is involved in the material or energy metabolism,indicating that all of these Gis have the function of metabolic island, and metabolic island is indispensable in cell survival.
Symbiosis island was first identified in Mesorhizobium loti ICMP3153 which contained P4 integrase and located in the 3’end of tRNA-Phe[14]. We observe that all of these Gis have homologous fragments with other species except the short ISL15 through blastP, as shown in Fig.1 A, and the degree of homology is gamma-proteobacteria, bacteroidetes,thermotogae, deinococcus-thermus, euryarchaeota,crenarchaeota, and virus in turn, indicating that a number of horizontal gene transfers happen in Syn. PCC 6803 and a lot of foreign genes are accepted. In addition, it shows that Syn. PCC 6803 is easier to get genetic material from distantly related species, and the traditional family tree is not applicable for the analysis of horizontal gene transfer in bacteria, because their genomes are the combination of genetic material of themselves and other species. However,we can still explore the lineage of Syn. PCC 6803 because the accepted genes are not included in the core gene pool and they may be lost again at any time. If not, they will be kept for offspring and have some functions. In all Gis, only ISL7 has a fragment with virus which locates in intergenic region, as shown in Fig. 1 B. We speculate that the selective pressure of virus increases the diversity of Gis and reduces the actual population of host in order to form symbiotic relationship under the natural condition.
Fitness islands are suggested to increase the fitness of the recipient bacteria, and they should confer new properties which enhance the adaptation capacity. Fitness islands were divided into different subsets depending on the life-style of host by Jörg Hacker and his colleagues[15].They pointed out that fitness islands which help bacteria to live can be called “ecological islands”, help bacteria to persist saprophyte can be called “saprophytic islands”, help bacteria to positively interact with their hosts can be called“symbiosis islands”, and help bacteria to participate in the induction of lesions can be called “pathogenicity islands”.We can see that fitness islands are not clearly distinguished from other types of Gis, and they will become other types when they have different functions. The gene products in our Gis are analyzed and we find that many genes involved in metabolism also take part in the bacterial adaption. That is, metabolic islands play an important part in stress response such as low temperature and low ph instead of fitness islands. In fact, the role of Gis is multiple. ISL7 has the function of both symbiosis island and metabolic island,and ISL16 has the function of both resistance island and fitness island.
We can see from our results that the Gis of Syn. PCC 6803 are diverse and multiple-functional, which to some extent can explain why Syn. PCC survivals millions of years and distributes so widely. Genomic islands, in essence, are not continuous DNA fragments. Bacteria obtain them through exchanging from homologous strains or other organisms in the same ecological habitat. The sources of Gis make them comprise an overarching family of elements with different functional life-styles. Initially genomic islands were found in E.coli and proposed the concept of pathogenicity islands (PAIs), but they have been extended to a much broader forms than PAIs only up to now, with variety of sizes and diversity of functions. As discussed in this paper, Gis are closely related with cell metabolism and evolution in addition to pathogenicity.Both the general (ISL1) and the unique (ISL15)characteristics of Syn. PCC 6803 can be found through the analysis of Gis.

Fig. 1. BlastP of Gis and other species. The results of ISL1, ISL7 and ISL15 are shown. Red arrow means the homologous region between Syn. PCC 6803 and virus.
We pay attention to the distribution of Yfr non-coding RNA family in Syn. PCC 6803 and find that different members have significant distinctions. For instance, Yfr1 exists in many strains while Yfr16 is just found in P.marinus str. MIT 9515. We compare four representative genomes of P. marinus MED4, P. marinus str. MIT 9515, P.marinus str. AS9601, and P. marinus SS120 to figure out the reason and the result is shown in Fig. 2. We can see that Yfr1 locates between trxA and guaB in all the four genomes. However, the sequences and wave shapes of Yfr1 in the former three genomes are almost the same while the last one is significantly different. The reason why Yfr1 is lost in P. marinus SS120 may be related to its special habitat. P. marinus SS120 has been isolated from greater depths as 150 m to 200 m and possesses numerous peculiarities with regard to other cyanobacteria like specific pigment complement and phycobilisome. Gene functions have greatly changed due to these unique cell structures in order to be adapted to the environment. This result is also accordant with the precious phylogenetic tree based on 16S rRNA. Yfr16 locates in homogenous blocks, but the position of Yfr16 is variable and special for only P. marinus str. MIT 9515. The sequence of this region is more prone to change through reorganization and horizontal transfer which causes uniqueness of Yfr16. In addition, we can see that both the location and the flanking blocks of the homologous region in P. marinus SS120 are different,which mainly attributes to its genomic modification and rearrangement for special habitat. Thus, we get conclusions that the environment is a significant factor which can not be ignored, the sequences at the boundary of the blocks and the breakpoints of genome rearrangement often change to be specific to a particular genome, and genome-wide comparative way is convenient and intuitive to detect these differences. The divergence in light absorption and metabolism makes their genomic rearrangement and evolution in different directions. Our studies confirm that the genome of cyanobacteria constantly remodel to adapt the environment during the long process of evolution.
Genome rearrangement perfectly answers the question why Yfr16 exists in certain strains. It also provides the evidence for the genome reconstruction and stress adaptation. Gis are part of flexible bacterial gene pool and are somewhere between 10 kb and 100 kb in length. Unlike the core gene pool that plays a role in basic cellular function and exhibits homology, the genes from the flexible gene pool often have the features of transferred elements and encode additional functions that are not essential for growth but provide advantages under stresses to increase fitness, which is important for their survival.
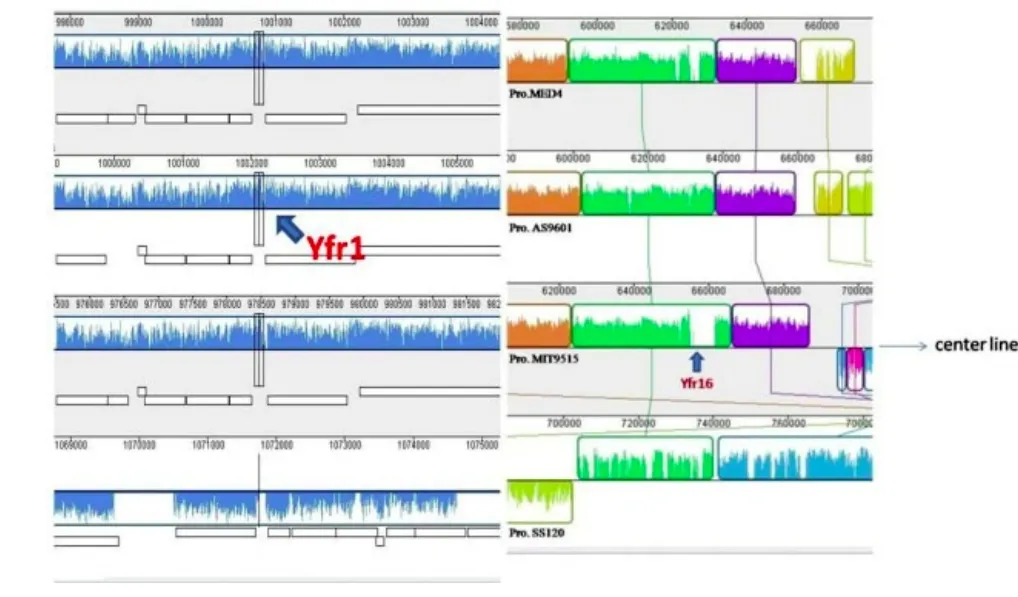
Fig. 2. Location of Yfr1 and Yfr16 in cyanobacteria. Homogenous blocks are labeled in the same color and linked with a line. Blocks above the center line are in the forward orientation, below in the reverse orientation. Regions outside blocks lack detectable homology, and inside each block is drawn a similarity profile of the genome sequence. Yfr16 locates at white areas that are not aligned and contain sequence elements specific to only Pro.MIT9515.
As seen from the results above that the formation of genomic islands is closely related with the horizontal transfer and recombination of foreign material. Dobrindt[16]et al. considered there are five steps to complete the whole process, and Gis are movable because they can be deleted and integrated into new chromosome. However, Ragan[2]et al. took a classification of Gis based on their mobility and found that most of Gis are not movable. The rest movable Gis are constituted of plasmid, prophage, conjugative transposon, and SXT element. It should be a dynamic equilibrium of integration and deletion at the initial stage of the Gi transfer. That is, integrase acts on the direct repeats of Gi which makes Gi circulated and dropped off from chromosome, but at the same time it is able to make Gi integrated back into chromosome. These special elements mentioned above are also important parameters for us to predict Gis. We analyze the results and find that some Gis have lost the mobility such as ISL1 because there are no movable elements and tRNA-associated integrated sites on them. While ISL11 has both of them, indicating it may be reintegrated and transferred again.
At present, genomic islands are thought to be more important for genome research and there are many different algorithms and software established. We can divide them into three categories. The first category is based on the sequence difference between Gis and chromosome such as GC content, dinucleotide bias, genetic code usage, and gene density. The second one is based on the structural characteristics of the Gis such as integrase, tRNA, tmRNA,and direct repeats. The third one is comparative genome that based on the location of Gis because they are sometimes unique and preferential on chromosome. It is a challenge to predict the Gis accurately because of their diversities of sequence and function, and there are defects in every method. For instance, the second category provides clear boundaries of Gis and finds the internal integrase, but only the Gis with tRNA or tmRNA site can be found and other types of integration site are ignored. In this article, we combine different categories of methods to improve the accuracy, which is the trend of the future researches.
4. Conclusions
In this article, we predict and analyze the Gis in Synechocystis sp. PCC 6803 by bioinfomatics. The results show that ISL1, ISL8 and ISL16 are homologous with many other bacteria. They involve in basic reactions and have a conservative evolution. On the contrary, ISL15 has a unique sequence and function only for Synechocystis sp.PCC 6803. Most of Gis play a role in genome rearrangement because they have lots of transposase.Moreover, we find that recombination and horizontal transfer of Gis is an important factor to affect the distribution of non-coding RNA. Our work contributes to a comprehensive understanding of genomic islands and their impact on genome of cyanobacteria and shows that Gis is important and is becoming a powerful tool to explore the unknown part of genome. In future study, we will verify these predictions next step through experiments.
[1] B. A. Whitton and M. Potts, The Ecology of Cyanobacteria:Their Diversity in Time and Space, Dordrecht: Kluwer Academic, 2000.
[2] M. Juhas, V. Der Meer, J. Roelof, M. Gaillard, R. M.Harding, D. W. Hood, and D. W. Crook, “Genomic islands:Tools of bacterial horizontal gene transfer and evolution,”FEMS Microbiology Reviews, vol. 33, no. 2, pp. 376-393,2009.
[3] E. Carniel, “The Yersinia high-pathogenicity island: An iron-uptake island,” Microbes and Infection, vol. 3, no. 7, pp.561-569, 2001.
[4] H. Karch, S. Schubert, D. Zhang, W. Zhang, H. Schmidt, T.Ölschläger, and J. Hacker, “A genomic island, termed high-pathogenicity island, is present in certain non-O157 Shiga toxin-producing Escherichia coli clonal lineages,”Infection and Immunity, vol. 67, no. 11, pp. 5994-6001,1999.
[5] Y. Yan, J. Yang, Y. Dou, M. Chen, S. Ping, J. Peng, W. Lu,W. Zhang, Z. Yao, and H. Li, “Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501,” Proc. of the National Academy of Sciences, vol. 105, no. 21, pp.7564-7569, 2008.
[6] A. Dufresne, M. Ostrowski, D. J. Scanlan, L. Garczarek, S.Mazard, B. P. Palenik, I. T. Paulsen, N. T. de Marsac, P.Wincker, and C. Dossat, “Unraveling the genomic mosaic ofa ubiquitous genus of marine cyanobacteria,” Genome Biol., vol. 9, no. 5, pp. 90-106, 2008.
[7] M. L. Coleman, M. B. Sullivan, A. C. Martiny, C. Steglich,K. Barry, E. F. DeLong, and S. W. Chisholm, “Genomic islands and the ecology and evolution of Prochlorococcus,”Science, vol. 311, no. 5768, pp. 1768-1770, 2006.
[8] S. Waack, O. Keller, R. Asper, T. Brodag, C. Damm, W. F.Fricke, K. Surovcik, P. Meinicke, and R. Merkl,“Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models,” BMC Bioinformatics, vol. 7, no. 1, pp. 142-154, 2006.
[9] W. Hsiao, I. Wan, S. J. Jones, and F. S. Brinkman,“IslandPath: Aiding detection of genomic islands in prokaryotes,” Bioinformatics, vol. 19, no. 3, pp. 418-420,2003.
[10] M. G. Langille, W. W. Hsiao, and F. S. Brinkman,“Evaluationof genomic island predictors using a comparative genomics approach,” BMC Bioinformatics, vol.9, pp. 329-339, Aug. 2008.
[11] A. E. Darling, B. Mau, and N. T. Perna, “progressiveMauve:multiple genome alignment with gene gain, loss and rearrangement,” PLoS One, vol. 5, no. 6, pp. e11147, 2010.[12] O. Gal-Mor and B. B. Finlay, “Pathogenicity islands: A molecular toolbox for bacterial virulence,” Cellular Microbiology, vol. 8, no. 11, pp. 1707-1719, 2006.
[13] B. Hochhut, K. Jahreis, J. W. Lengeler, and K. Schmid,“CTnscr94, a conjugative transposon found in enterobacteria,” Journal of Bacteriology, vol. 179, no. 7, pp.2097-2102, 1997.
[14] J. T. Sullivan and C. W. Ronson, “Evolution of rhizobia by acquisition of a 500-kB symbiosis island that integrates into a phe-tRNA gene,” Proc. of the National Academy of Sciences, vol. 95, no. 9, pp. 5145-5149, 1998.
[15] J. Hacker and E. Carniel, “Ecological fitness, genomic islands and bacterial pathogenicity,” EMBO Reports, vol. 2,no. 5, pp. 376-381, 2001.
[16] U. Dobrindt, B. Hochhut, U. Hentschel, and J. Hacker,“Genomic islands in pathogenic and environmental microorganisms,” Nature Reviews Microbiology, vol. 2, no.5, pp. 414-424, 2004.
 Journal of Electronic Science and Technology2014年2期
Journal of Electronic Science and Technology2014年2期
- Journal of Electronic Science and Technology的其它文章
- Perfect Reconstructable Decimated One-Dimensional Empirical Mode Decomposition Filter Banks
- Structure Analysis and Classification of Precipitation Cells by Fractal Geometry
- Adaptive Fourier Decomposition Based Time-Frequency Analysis
- Retrieval of High Resolution Satellite Images Using Texture Features
- Call for Papers Journal of Electronic Science and Technology Special Section on Special Section on Energy-Efficient Technologies
- Journal of Electronic Science and Technology Information for Authors