
LTF基因在胃癌细胞株BGC823中的表达及其与启动子甲基化的相关性
2014-06-13 10:53诸葛小菊陈仁聘黄燮林陈超卢的一黄智铭吴金明吴建胜
温州医科大学学报 2014年6期
诸葛小菊,陈仁聘,黄燮林,陈超,卢的一,黄智铭,吴金明,吴建胜
(1.温州医科大学附属第一医院 消化内科,浙江 温州 325015;2.温州医科大学 仁济学院,浙江温州 325035)
胃癌的发病率高居恶性肿瘤的前4位,其发病过程受到多基因、多因素的影响[1]。越来越多的证据表明抑癌基因的失活在肿瘤发生发展中起重要作用。在胃癌癌变早期,抑癌基因的表观遗传学改变占有重要的地位,尤其是抑癌基因启动子的异常甲基化[2]。
Lactotransferrin(LTF)基因位于人类染色体3p21.3区域,其在鼻咽癌、肺癌、前列腺癌等多种肿瘤及相应的肿瘤细胞株呈现表达降低或缺失的状态,有研究[3-6]显示这与该基因启动子区域的异常甲基化高度相关。5-Aza-2’-deoxycytidine(5-aza-CdR)能结合DNA甲基转移酶I并使其失活,逆转抑癌基因的高甲基化状态,重新激活基因表达。而目前对于LTF基因在胃癌内的研究尚少,因此本研究选择胃癌细胞株BGC823,观察LTF基因的表达和其启动子甲基化状态,旨在研究胃癌细胞株内LTF基因表达水平和启动子异常甲基化的相关性,为胃癌临床诊断治疗提供一个新的可能靶点。
1 材料和方法
1.1 材料 人胃腺癌细胞株BGC823购于中国上海科学院细胞库;胎牛血清购于杭州四季青生物工程材料有限公司;PRMI-1640、PBS、青/链霉素100 U/mL购于Gibco公司;5-aza-CdR(Sigma公司),用培养基充分溶解配成适量浓度的母液,分装后存于-80 ℃备用;TRIzol购于Invitrogen公司;反转录试剂盒、real-time PCR试剂盒(SYBR®Green Re altime PCR Master Mix)购于日本TOYOBO公司;亚硫酸氢钠修饰试剂盒购于美国ZYMO公司;Ex Taq HS DNA聚合酶、pMD 19-T Vector购于日本TaKaRa公司;抗体anti-Lactoferrin购于美国Abcam公司(77 kDa,1:1 000#ab10110;Abcam,Cambridge,UK);β-actin antibody购于中国碧云天(42 kDa,1:1 000,Beyotime,中国上海)。荧光定量PCR仪为BIO-RAD(型号CFX96)。
1.2 方法
1.2.1 细胞的培养和5-aza-CdR药物处理:BGC82 3细胞用含10%胎牛血清的PRMI-1640培养液放置在37 ℃,5% C02的细胞培养箱内培养。以1×105均匀铺入六孔板内,生长过夜。饥饿处理16 h,然后给予0 μmol/L(对照组)、2 μmol/L、10 μmol/L的5-aza-CdR药物处理,24 h换液一次,培养5 d后处理细胞。
1.2.2 real-time PCR检测药物干预前后LTF基因mRNA表达:取对照组和药物处理组的回收细胞,用TRIzol提取细胞总RNA。约取1 μg总RNA根据反转录试剂盒说明书反应得到20 μL体系的cDNA。以得到cDNA为模板,进行real-time PCR反应。引物由上海生工公司设计并合成,用GAPDH作为内参,引物序列分别为上游5’-GACTCATGACCACAGTCCATGC-3’,下游5’-AGAGGCAGGGATGATGTTCTG-3’,扩增产物长度113 bp。LTF引物序列分别为:上游5’-TACAAATC CCAACAAAGCAGTG-3’,下游5’-CTTCCACAGGTCTATCC ACACA-3’,产物大小约58 bp。20 μL PCR体系包括:Realtime PCR Master Mix 10 μL,Plus solution 2 μL,PCR Forward Primer(10 μmol/L)1.2 μL,PCR Reverse Primer(10 μmol/L)1.2 μL,模板cDNA 2 μL,ddH2O 3.6 μL。反应条件是95 ℃预变性30 s,95 ℃ 5 s、62 ℃ 10 s、72 ℃ 15 s进行40个循环。扩增结束进行熔解曲线分析来辨别是否有非特异性扩增。实验重复3次。数据 用2-ΔΔCt法分析。
1.2.3 Western blot法检测药物干预前后LTF基因蛋白表达:回收对照组和药物处理组细胞,加入适量的蛋白抽提cell lysis buffer(每孔50μg)裂解细胞,然后用BCA法测定蛋白质的浓度。用12% SDS-聚丙烯酰胺凝胶进行电泳,将蛋白用半干式电转移法转至硝酸纤维膜上,用5%脱脂牛奶TBST封闭硝酸纤维膜,室温下摇2 h。与一抗anti-Lactoferrin(77 kDa)结合(以TBS稀释,浓度依抗体而定),4 ℃摇过夜,TBST洗4次,每次5 min,用β-actin antibody(42 kDa)作为内参。加入二抗(以TBS稀释,浓度依抗体而定),室温摇1 h,TBST洗4次,每次5 min。用ECL试剂盒进行化学发光检测。X线片压片、显影、定影。
1.2.4 亚硫酸氢盐修饰DNA和BSP反应:取对照组和药物处理组的回收细胞,按DNA提取试剂盒提取胃癌细胞株的基因组DNA。然后按甲基化修饰试剂盒进行亚硫酸氢钠修饰,使未甲基化的胞嘧啶(C)转化成胸腺嘧啶(T),而甲基化的Cm不变。以修饰后基因组DNA为模板,用Ex Taq HS DNA聚合酶进行LTF基因启动子扩增。LTF特异性引物参考文献[5],分别是上游5’-TTGAGATTAGAGTTGGGATAGGG-3’,下游5’-CCCCCAAACACCTACACTCA-3’,产物长度为397 bp。其反应体系为50 μL,包括:TaKaRa Taq HS 0.25 μL,10×PCR Buffer 5 μL,dNTP Mixture 4 μL,上游引物(10 μmol/L)1 μL,下游引物(10 μmol/L)1μL,模板DNA 1 μL(≤50 ng),ddH2O 37.75 μL。反应条件为95 ℃变性15 min,94 ℃ 30 s、56℃ 30 s、72 ℃ 30 s进行35次循环,然后72 ℃延伸7 min。将获得的PCR产物用1.5%琼脂糖凝胶电泳,凝胶图像分析系统记录数据。
1.2.5 挑选阳性克隆和测序:于紫外灯下切取含目的DNA的条带,按DNA凝胶回收试剂盒说明书对PCR产物进行回收纯化。取回收纯 化的DNA 4.5 μL,与pMD 19-T Vector 0.5 μL、solution I 5 μL配成10 μL体系,轻轻混匀后16 ℃连T载体12 h。取2.5 μL连接产物与25 μL的大肠杆菌感受态冰浴30 min,42 ℃水浴90 s,冰上放置3 min,加入不含氨苄青霉素的LB培养基600 μL,37 ℃,250 r/min振摇45 min,离心将菌液浓缩为15 μL,并涂于氨苄(100 μg/mL)抗性的LB平板上,先正面向上放置平板1 h风干,翻转倒置平板37 ℃培养过夜,12~16 h后即可长出菌落。随机挑选15个单克隆菌落,放在500 μL LB液体培养基(含有氨苄青霉素100 μg/mL),摇浑浊菌液。用普通PCR鉴定阳性重组子,取1 μL摇浑的菌液做PCR模板,2×Taq酶Mix 5 μL,上下游通用引物各0.2 μL,ddH2O 3.6 μL构成10 μL PCR体系进行扩增反应。用1.5%琼脂糖凝胶电泳鉴定结果。最后送检10个克隆于上海生工公司测序。
1.3 统计学处理方法 采用SPSS13.0统计软件。real-time PCR结果和总体甲基化率组间比较采用单因素方差分析,组间两两比较采用t检验。CpG位点甲基化率比较用行×列表卡方检验。P<0.05为差异有统计学意义。
2 结果
2.1 胃癌细胞株BGC823 LTF基因的mRNA 表达 不同浓度5-aza-CdR处理胃癌细胞株BGC823后,药物处理的两组其mRNA表达较对照组明显增加,差异有统计学意义(P<0.05),但药物处理组间差异无统计学意义(P>0.05)。结果如图1A所示。我们用Western blot方法检测药物干预前后LTF基因蛋白表达,结果如图2所示,对照组LTF表达低,两个不同浓度的药物组较对照组表达明显增加。
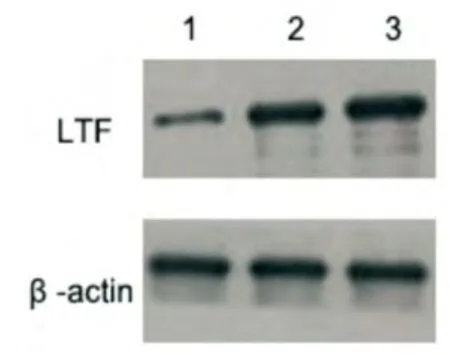
图2 5-aza-CdR药物处理前后LTF的蛋白表达
2.2 胃癌细胞株BGC823药物处理前后LTF基因甲基化情况 BSP结果电泳图如图3所示,目的条带397 bp清晰。LTF基因连接T载体后挑选阳性克隆电泳结果如图4所示,表明所选阳性单克隆概率高,达83.3%(5/6)。

图3 LTF基因的BSP法电泳图结果

图4 LTF甲基化后目的DNA连T载体挑选阳性克隆电泳图
BGC823被不同浓度5-aza-CdR处理后LTF其总体甲基化率统计见图1B,其中对照组表明胃癌 细胞株BGC823明显高甲基化,而药物处理后的两组启动子甲基化情况被逆转,且两个药物处理组和对照组比差异有统计学意义(甲基化率对照组为53.6%,药物浓度2μmol/L组为27.9%,浓度10μmol/L组为35.0%)(P<0.05),但药物处理组间差异无统计学意义(P>0.05)。LTF基因启动子区14个CpG位点甲基化情况如图5显示,其中3、4、5、8、10、11、12、13、14号CpG位点甲基化程度超过50%,呈高甲基化状态。而用5-aza-CdR处理后,与对照组相比,14号位点甲基化情况几乎没有变化,3、8这两个CpG位点甲基化率较对照组明显降低,差异有统计学意义(P<0.05)。
3 讨论
胃癌的发生机制是涉及多因素多步骤的一个过程,主要包括遗传性因素和表观遗传学因素改变的堆积,而后者主要包括启动子区甲基化、杂合性丢失、组蛋白修饰三方面。越来越多的研究表明DNA启动子区甲基化在肿瘤的发生发展过程中占有重要的地位。Lund等[7]认为DNA甲基化在某种时候甚至超越基因缺失和基因突变,是抑癌基因失活的唯一机制。DNA甲基化是一可逆过程,5-aza-CdR作为一种甲基化转移酶,可以成功逆转DNA的甲基化程度,使得原先因甲基化而失活的基因能够重新表达或表达增强,恢复正常的细胞调控功能。这提示去甲基化剂治疗能够逆转基因的甲基化状态,具有一定的应用前景。
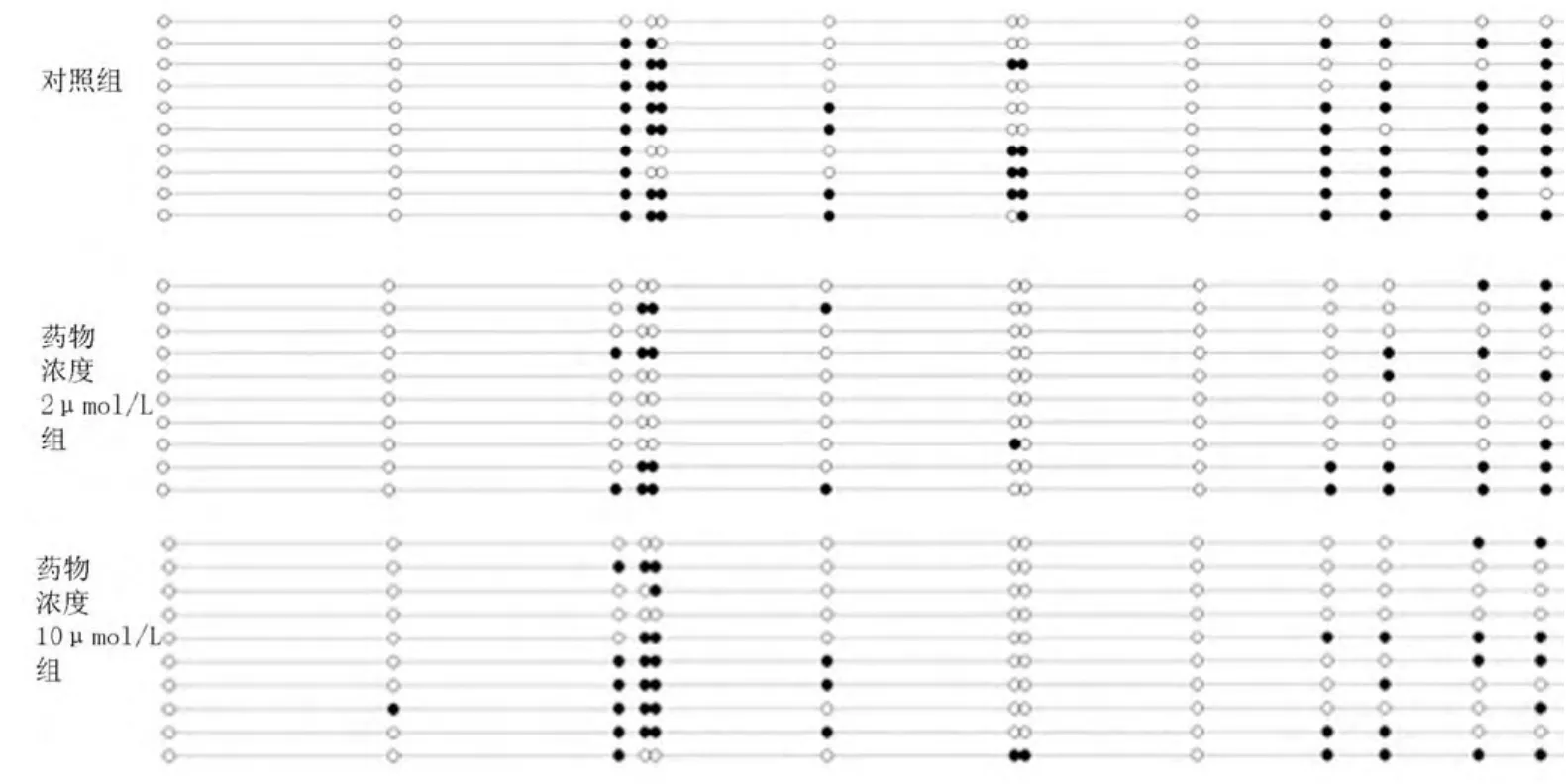
图5 5-aza-CdR药物处理前后LTF基因启动子区14个CpG位点甲基化情况
研究发现人体染色体3p21.3区域存在一群高密度分布的肿瘤抑制基因簇(TSGs),该区域的抑癌基因在肺癌、乳腺癌、肾癌和其他肿瘤中常常表达缺失[8]。LTF基因,亦是其中重要一员,编码乳铁蛋白(lactoferrin,LF),其主要存在于哺乳动物的乳液、唾液、泪液、精液等外分泌物中,能参与体内铁代谢、抗菌、细胞生长和分化、免疫调节和抗炎症反应、抗氧化反应等多种生物学效应[9-10]。近来研究发现,LF在抑制肿瘤生长、转移,促进肿瘤细胞凋亡方面也有重要作用[11]。
Iijima等[5]报道LTF基因在59%(16/27)小细胞肺癌细胞株,77%(33/43)非小细胞肺癌细胞株,54%(7/33)原发性非小细胞肺癌内表达缺失,且与DNA甲基化高度相关。研究[6]发现LTF基因mRNA在74%前列腺癌患者中呈低表达,而且LTF相对高表达的前列腺癌患者有更好的预后,这反映LTF在前列腺癌患者中有保护性的功能。DNA去甲基化剂地西他滨(decitabine)能使LTF基因在前列腺癌细胞株LAPC4中被强激活表达。LTF基因在多种肿瘤内表现为一种有潜力的抑癌基因,其失活机制和DNA启动子区高度甲基化相关。本研究观察药物处理组和对照组发现,随着药物处理组DNA甲基化率的下降,其LTF mRNA表达增加,这说明LTF基因在胃癌里的表达是可以被逆转的,而且其机制和DNA甲基化高度相关。但是两个药物处理组间的甲基化率和mRNA表达差异无统计学意义,这可能和5-aza-CdR对BGC823的去甲基化作用达到平台期相关。重亚硫酸盐修饰DNA测序法,是公认的DNA甲基化分析的金标准。本研究用该方法检测发现,低分化型胃癌细胞株BGC823启动子区甲基化程度高达53.6%,该段DNA启动子区含有14个CpG岛,CpG岛是一段长约1 kb的富含饰CpG和GpC序列的DNA结构。进一步分析发现3、4、5、8、10、11、12、13、14号CpG位点甲基化程度超过50%,呈高甲基化状态,而用5-aza-CdR处理后,14号位点甲基化率几乎没有变化,而3、8这两个CpG位点甲基化率较对照组下降,差异有统计学意义(P<0.05),猜测这两个位点和LTF基因在胃癌中的表达缺失及逆转其表达可能相关。
大量研究证明LF在体内外实验中有抗肿瘤作用[12-14],但是其机制目前并不明确。LF能以影响细胞周期的正常运转来抑制肿瘤细胞的增殖,Zhang等[4]用LTF转染鼻咽癌细胞株后,发现,细胞停滞在G1期,而S期、G2-M期细胞减少。有研究[15]认为LF能以调节细胞周期相关蛋白和抑癌基因(p53和Rb)的表达来影响细胞周期的整个进程。但是亦有报道持不同意见,认为LF补给治疗能调节MAPK(mitogenactivated protein kinase)通路却不会影响p53的表达[14]。体外实验证明注射LF能抑制小鼠体内移植实体瘤的大小,Fujita等[16]发现LF在小鼠结肠癌发展过程中,激活Fas信号通路导致结肠癌细胞的凋亡。当然,对于LTF基因在胃癌内的抑癌机制是否涉及以上几种,需更多的实验研究。
DNA甲基化作为表观遗传学重要机制之一,其本身优势在于不涉及基因序列的改变就可以调控基因的表达,并且能够被甲基化转移酶抑制剂逆转。5-aza-CdR,作为一种甲基化转移酶,已成功应用于白血病的临床治疗[17],这对抑癌基因甲基化异常相关的肿瘤来说是个成功的例子。针对LTF基因在胃癌内表达和基因甲基化机制的关联,提示我们未来可能通过甲基化转移酶抑制剂来进行肿瘤的预防和早期治疗,当然这还需要进一步的实验及临床研究。
[1]Jemal A, Bray F, Center MM, et al. Global cancer statistics[J]. CA Cancer J Clin, 2011, 61(2): 69-90.
[2]Choi IS, Wu TT. Epigenetic alterations in gastric carcinogenesis[J]. Cell Res, 2005, 15(4): 247-254.
[3]Yi HM, Li H, Peng D, et al. Genetic and epigenetic alterations of LTF at 3p21.3 in nasopharyngeal carcinoma[J].Oncol Res, 2006, 16(6): 261-272.
[4]Zhang H, Feng X, Liu W, et al. Underlying mechanisms for LTF inactivation and its functional analysis innasopharyngeal carcinoma cell lines[J]. J Cell Biochem, 2011,112(7): 1832-1843.
[5]Iijima H, Tomizawa Y, Iwasaki Y, et al. Genetic and epigenetic inactivation of LTF gene at 3p21.3 in lung cancers[J]. Int J Cancer, 2006, 118(4): 797-801.
[6]Shaheduzzaman S, Vishwanath A, Furusato B, et al. Silencing of Lactotransferrin expression by methylation in prostate cancer progression[J]. Cancer Biol Ther, 2007, 6(7): 1088-1095.
[7]Lund AH, van Lohuizen M. Epigenetics and cancer[J].Genes Dev, 2004, 18(19): 2315-2335.
[8]Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster[J]. Oncogene, 2007, 26(52):7283-7301.
[9]Hayakawa T, Jin CX, Ko SB, et al. Lactoferrin in gastrointestinal disease[J]. Intern Med, 2009, 48(15): 1251-1254.
[10]Valenti P, Antonini G. Lactoferrin: an important host defence against microbial and viral attack[J]. Cell Mol Life Sci, 2005, 62(22): 2576-2587.
[11]Artym J. [Antitumor and chemopreventive activity of lactoferrin][J]. Postepy Hig Med Dosw (Online), 2006, 60: 352-369.
[12]Tsuda H, Kozu T, Iinuma G, et al. Cancer prevention by bovine lactoferrin: from animal studies to human trial[J]. Biometals, 2010, 23(3): 399-409.
[13]Yamada Y, Sato R, Kobayashi S, et al. The antiproliferative effect of bovine lactoferrin on canine mammary gland tumor cells[J]. J Vet Med Sci, 2008,70(5): 443-448.
[14]Zhou Y, Zeng Z, Zhang W, et al. Lactotransferrin: a candidate tumor suppressor-Defi cient expression in humannasopharyngeal carcinoma and inhibition of NPC cell proliferation by modulatingthe mitogen-activated protein kinase pathway[J]. Int J Cancer, 2008, 123(9): 2065-2072.
[15]Son HJ, Lee SH, Choi SY. Human lactoferrin controls the level of retinoblastoma protein and its activity[J]. Biochem Cell Biol, 2006, 84(3): 345-350.
[16]Fujita K, Matsuda E, Sekine K, et al. Lactoferrin enhances Fas expression and apoptosis in the colon mucosa ofazoxymethane-treated rats[J]. Carcinogenesis, 2004, 25(10): 1961-1966.
[17]Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine inhigher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia[J]. Blood, 2007, 109(1): 52-57.
猜你喜欢
中成药(2018年3期)2018-05-07
中国男科学杂志(2016年9期)2016-03-20
中国医疗美容(2015年1期)2015-07-12
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国医药导报(2015年27期)2015-02-28
癌变·畸变·突变(2015年3期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
遗传(2014年3期)2014-02-28
