
Activities of nicotinic acetylcholine receptors modulate neurotransmission and synaptic architecture
2014-06-01 09:12AkiraOdaHidekazuTanaka
中国神经再生研究(英文版) 2014年24期
Akira Oda, Hidekazu Tanaka
1 CNS Drug Discovery Unit, Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, 2-26-1, Muraoka-higashi, Fujisawa, Kanagawa 251-8555, Japan
2 Laboratory of Pharmacology, Department of Biomedical Sciences, College of Life Sciences, Ritsumeikan University, 1-1-1, Noji-higashi, Kusatsu, Shiga 525-8577, Japan
Activities of nicotinic acetylcholine receptors modulate neurotransmission and synaptic architecture
Akira Oda1, Hidekazu Tanaka2
1 CNS Drug Discovery Unit, Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, 2-26-1, Muraoka-higashi, Fujisawa, Kanagawa 251-8555, Japan
2 Laboratory of Pharmacology, Department of Biomedical Sciences, College of Life Sciences, Ritsumeikan University, 1-1-1, Noji-higashi, Kusatsu, Shiga 525-8577, Japan
The cholinergic system is involved in a broad spectrum of brain function, and its failure has been implicated in Alzheimer’s disease. Acetylcholine transduces signals through muscarinic and nicotinic acetylcholine receptors, both of which in fl uence synaptic plasticity and cognition. However, the mechanisms that relate the rapid gating of nicotinic acetylcholine receptors to persistent changes in brain function have remained elusive. Recent evidence indicates that nicotinic acetylcholine receptors activities affect synaptic morphology and density, which result in persistent rearrangements of neural connectivity. Further investigations of the relationships between nicotinic acetylcholine receptors and rearrangements of neural circuitry in the central nervous system may help understand the pathogenesis of Alzheimer’s disease.
cholinergic system; nicotinic acetylcholine receptors (nAChRs); Alzheimer’s disease (AD); synaptic transmission; synaptic plasticity; synaptic morphology; dendritic spine remodeling; cognition
Funding: Tanaka H is supported by the Takeda Science Foundation and JSPS KAKENHI Grant Number 19590247.
Oda A, Tanaka H. Activities of nicotinic acetylcholine receptors modulate neurotransmission and synaptic architecture. Neural Regen Res. 2014;9(24):2128-2131.
Nicotinic receptors in the central nervous system (CNS) are candidate targets for the treatment of dementia
Dementia is one of the biggest global public health challenges. Accordingly, over 40 million people suffer from dementia globally, and this number is expected to double by 2030 and more than triple by 2050 (Alzheimer’s disease International, World Alzheimer Report 2014). Alzheimer’s disease (AD) is the most common type of dementia, and accounts for an estimated 60 to 80% of cases.
Loss of cholinergic neurons is a pathological hallmark of AD, and is also assessed in human imaging studies (Nordberg et al., 2010). Cholinergic neurons of the nucleus basalis of Meynert and the medial septum innervate the cerebral cortex and the hippocampus, respectively, and are involved in cognition. Hence enhancement of cholinergic transmission with acetylcholinesterase inhibitors ameliorates cognitive deficits in AD patients (Rogers et al., 1998). Consistently, deprivation of cholinergic transmission by either genetic disruption of acetylcholine receptors or denervation of cholinergic nerve fi bers severely impairs learning and memory in animal experiments (Champtiaux and Changeux, 2004; Wess, 2004).
The cholinergic system also plays pivotal roles in other psychiatric disorders (Scarr et al., 2013). For example, an imbalance between cholinergic and dopaminergic systems contributes to the pathophysiology of schizophrenia, and adjunctive use of acetylcholinesterase inhibitors alleviates visual hallucinations (Patel et al., 2010). In contrast, acetylcholine receptor antagonism ameliorates depressive symptoms in major depression and bipolar disorders (Drevets et al., 2013).
The neurotransmitter acetylcholine transduces signals through muscarinic and nicotinic acetylcholine receptors. Nicotinic acetylcholine receptor (nAChR) comprises five subunit polypeptides. Brain-type nAChRs consist of heteromeric combinations of α2–10 and β2–4 subunits, as well as α7 homopentamers; α4β2*and α7 nAChRs are predominantly expressed in the brain (an asterisk indicates that the receptor may include other subunits than the described ones). In the pathophysiology of AD, amyloid β (Aβ), the peptide in senile plaques characteristic of AD, binds with high affinity to α7 nAChRs, and impairs their channel functions. The selective vulnerability of nAChR‐expressing neurons to Aβ toxicity may lead to the failure of cholinergic neurotransmission and cognitive dysfunctions in AD (Dineley, 2007).
Several nAChR‐targeting compounds have been developed to relieve neuropsychiatric symptoms in AD patients. However, clinical trials of compounds that target α4β2*and α7 nAChRs (Table 1) have produced discouraging results (see review, Hurst et al., 2013). For example, varenicline, a partial agonist of α4β2*nAChR, is the only clinically available nAChR‐target‐ing drug, and was approved for smoking cessation, but failed to improve cognition in AD patients (Kim et al., 2014).
The failure of nAChR‐targeting therapy for AD may be at‐tributed to several reasons. First, the expression of nAChRs is down‐regulated in the brains of AD patients (Nordberg et al., 2010). Moreover, the expression of α4β2*nAChRs is decreased in these patients, especially in affected brain areas, and appears to be correlated with the severity of cog‐nitive impairment and the amount of amyloid deposition (Sabri et al., 2008; Okada et al., 2013). Conditions relating to α7 nAChRs have been more controversial, with reports of decreased (Burghaus et al., 2000), stable (Ikonomovic et al., 2009), and increased (Nordberg, 2001) α7 nAChR expression in AD patients. Second, tested compounds have insufficient potencies, and under conditions of decreased nAChR subunit expression, these reportedly activate only a small fraction (about 10%) of nAChRs (Kim et al., 2014). Third, adverse effects of nAChR-targeting compounds, such as gastrointestinal and CNS-related side effects (Hurst et al., 2013), may limit therapeutic doses of compounds. To circumvent these side effects, partial agonists for nAChRs have been developed with the expectation of broader therapeutic windows than full agonists (Table 1). Nonetheless, a better understanding of the relationships between nAChRs and plasticity or rearrangements of neural circuitry may be required for the successful treatment of AD.

Table 1 Development of nicotinic acetylcholine receptor (nAChR) ligands for the treatment of Alzheimer’s disease and cognitive disorders
Nicotinic activity modulates plasticity and various synaptic transmissions
In addition to acetylcholine, major neurotransmitters inthe CNS include glutamate, GABA, dopamine, serotonin, and noradrenaline. These molecules are involved in various physiological processes, such as learning, memory, mood, appetite, anxiety, and fear. Corresponding synaptic transmissions mediated by these neurotransmitters are known to be modulated by nAChRs, which are localized to pre- and postsynaptic membranes, and have been demonstrated in multiple brain regions including the cerebral cortex, striatum, hippocampus, thalamus, and cerebellum (Gotti et al., 2006).
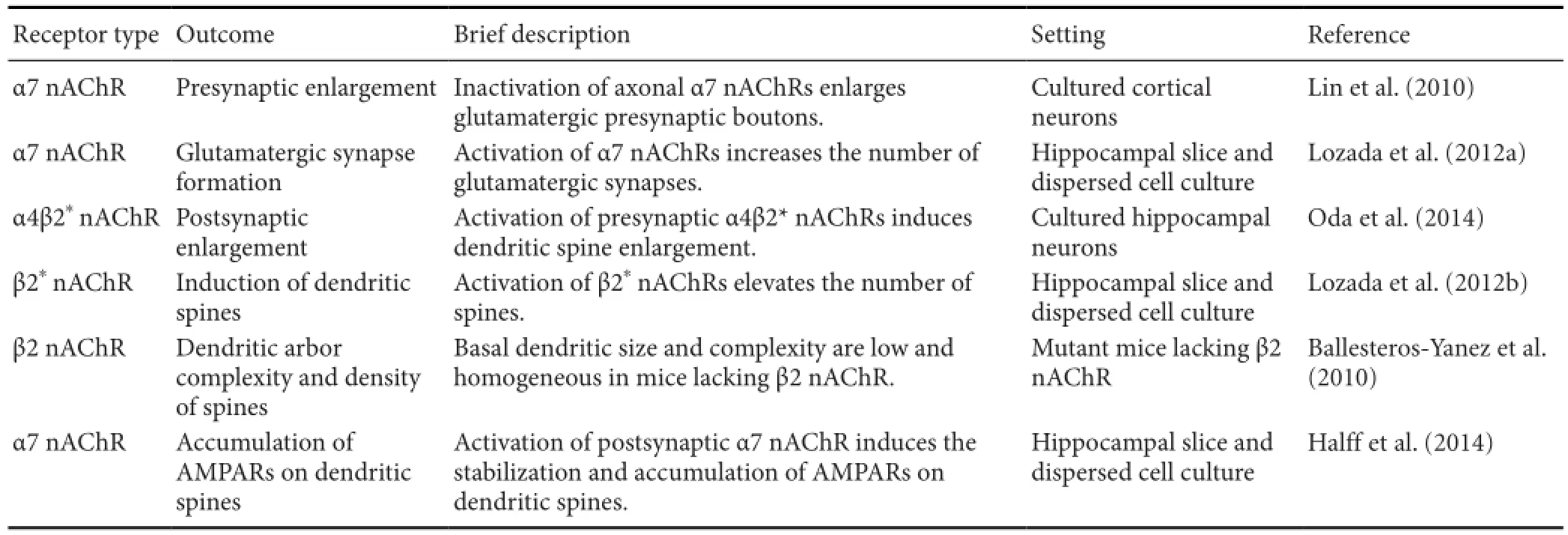
Table 2 Nicotinic acetylcholine receptors modulate synaptic architecture
Presynaptically, changes in the probabilities of transmitter release from synaptic vesicles have been reported (Placzek et al., 2009). Among these, spontaneous release of glutamate, as re fl ected in the frequency of miniature excitatory postsynaptic currents, is enhanced by the activation of presynaptic nAChRs. In addition, stimulus-dependent glutamate release is facilitated by the activation of nAChRs (Radcliffe et al., 1999; Rousseau et al., 2005). The mechanism underlying these processes reportedly involves increased presynaptic calcium levels following the actions of nAChRs (Placzek et al., 2009).
Long-term potentiation (LTP), a well-known form of synaptic plasticity in glutamatergic synapses, is modulated by postsynaptic nAChRs. Postsynaptic nAChR activities in pyramidal neurons boost the induction of LTP in hippocampal slices (Ji et al., 2001). Induction of LTP depends on the precise timing of presynaptic action potentials relative to postsynaptic depolarization (Dan and Poo, 2004). Coincidence or precedence of mild presynaptic stimulation with nAChR-mediated action potentials also leads to the induction of LTP (Ge and Dani, 2005; Gu and Yakel, 2011).
The excitability of GABAergic interneurons is reportedly modulated by postsynaptic nAChRs, and release of GABA from interneurons in turn modulates the excitability of pyramidal neurons, which further in fl uence the tone of neural circuits (Placzek et al., 2009). Local treatments of interneurons with acetylcholine activate nAChRs, resulting in the burst fi ring of action potentials of interneurons (Frazier et al., 1998; Alkondon et al., 1999).
Dual modulatory mechanisms of synaptic transmission through pre- and postsynaptic nAChRs may in fl uence brain activity and behavior. Thus, the impairment of nAChR functions in AD results in the failure of broad spectrum of synaptic transmissions.
Activation of nAChRs leads to rearrangements of synaptic architecture
Although nicotine rapidly modulates synaptic transmissions through nAChRs, exposure to nicotine produces persistent cravings for nicotine after long periods of abstinence (Dani and De Biasi, 2001). However, the mechanisms that relate rapid gating of nAChR to persistent changes in brain function have remained elusive. Recent evidence indicates that nAChR activities affect synaptic morphology and density, and neurotransmitter receptor distributions (Table 2).
Excitatory synapses in the CNS are usually formed on dendritic spines, which are small protrusions on dendrites. Dendritic spines receive glutamatergic inputs from apposing presynaptic termini, and are morphologically modulated by released glutamate (Okamura et al., 2004; McKinney, 2010). Enlargement and shrinkage of these spines are associated with LTP and long-term depression, respectively (Tada and Sheng, 2006). Although neuronal α4β2*and α7 nAChRs have been shown to modulate glutamatergic neurotrans‐mission, little is known of the accompanying alterations of synaptic architecture. In a recent study, nicotine was shown to induce dendritic spine enlargement through the activa‐tion of α4β2*nAChRs, which localize to presynaptic termini as indicated by super‐resolution structured illumination microscopy (Oda et al., 2014). Moreover, nicotine induces acute de novo glutamatergic synaptogenesis via β2*nAChRs in vitro and in vivo (Lozada et al., 2012a). In the absence of β2 nAChRs, the density of spines is reduced (Ballesteros‐Ya‐nez et al., 2010) and excitatory synaptic loci shift from spines to dendritic shafts (Lozada et al., 2012a). These observations are consistent with the hypothesis that activation of α4β2*nAChRs in fl uences the modulation and maintenance of syn‐aptic architecture.
Chronic exposure to nicotine promotes glutamatergic synaptogenesis, which is mediated by α7 nAChRs (Lozada et al., 2012b). Moreover, signi fi cant reductions in numbers of glutamatergic synapses have been observed in α7 nA‐ChR knockout mice, resulting in imbalances of excitatory/ inhibitory inputs to neurons (Lozada et al., 2012b). In con‐trast, chronic inactivation of α7 nAChRs leads to increased numbers of presynaptic boutons by enhancing presynaptic N‐methyl‐D‐aspartate receptor (NMDAR) function (Lin etal., 2010), indicating that the desensitization of α7 nAChRs also contributes to changes in synaptic architecture.
Recently, the molecular mechanisms of nAChR‐mediated modulation of synaptic function were demonstrated using super ecliptic pHluorin‐tagged α‐amino‐3‐hydroxy‐5‐meth‐yl‐4‐isoxazolepropionic acid receptors (AMPARs) (Ashby et al., 2004); surface insertion and internalization of AMPARs follows nicotinic stimulation (Hal ff et al., 2014). Postsynaptic morphological plasticity is oThen accompanied by changes in numbers of cell surface glutamate receptors, and stimulation of postsynaptic α7 nAChRs results in stabilization and accumulation of GluA1-containing AMPARs on dendritic spines (Halff et al., 2014). Hence, insertion and removal of synaptic AMPARs may be related to nicotine-induced modi fi cations of synaptic structure and function, as was the case with glutamate-induced plasticity (Kessels and Malinow, 2009).
Current treatments for AD are limited to symptomatic relief and fail to prevent disease progression. Thus, AD treatments that produce persistent rearrangements of neural connectivity are required and may substantially ameliorate the pathogenesis of AD. Further investigations of the relationships between nAChR and persistent rearrangements of neural circuitry in the CNS may elucidate therapeutic targets within the CNS cholinergic system.
Con fl icts of interest: Oda A is an employee of Takeda Pharmaceutical Company Limited. The authors declare no competing financial interests.
Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX (1999) Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci 19:2693-2705.
Ashby MC, Ibaraki K, Henley JM (2004) It’s green outside: tracking cell surface proteins with pH-sensitive GFP. Trends Neurosci 27:257-261.
Ballesteros-Yanez I, Benavides-Piccione R, Bourgeois JP, Changeux JP, DeFelipe J (2010) Alterations of cortical pyramidal neurons in mice lacking high-affinity nicotinic receptors. Proc Natl Acad Sci U S A 107:11567-11572.
Burghaus L, Schutz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, Lindstrom J, Schroder H (2000) Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res 76:385-388.
Champtiaux N, Changeux JP (2004) Knockout and knockin mice to investigate the role of nicotinic receptors in the central nervous system. Prog Brain Res 145:235-251.
Dan Y, Poo MM (2004) Spike timing-dependent plasticity of neural circuits. Neuron 44:23-30.
Dani JA, De Biasi M (2001) Cellular mechanisms of nicotine addiction. Pharmacol Biochem Behav 70:439-446.
Dineley KT (2007) Beta-amyloid peptide--nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front Biosci 12:5030-5038.
Drevets WC, Zarate CA, Jr., Furey ML (2013) Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry 73:1156-1163.
Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV (1998) Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci 18:1187-1195.
Ge S, Dani JA (2005) Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. J Neurosci 25:6084-6091.
Gotti C, Zoli M, Clementi F (2006) Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 27:482-491.
Gu Z, Yakel JL (2011) Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron 71:155-165.
Halff AW, Gomez-Varela D, John D, Berg DK (2014) A novel mechanism for nicotinic potentiation of glutamatergic synapses. J Neurosci 34:2051-2064.
Hurst R, Rollema H, Bertrand D (2013) Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther 137:22-54.
Ikonomovic MD, Wecker L, Abrahamson EE, Wuu J, Counts SE, Ginsberg SD, Mufson EJ, Dekosky ST (2009) Cortical alpha7 nicotinic acetylcholine receptor and beta-amyloid levels in early Alzheimer disease. Arch Neurol 66:646-651.
Ji D, Lape R, Dani JA (2001) Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 31:131-141.
Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61:340-350.
Kim SY, Choi SH, Rollema H, Schwam EM, McRae T, Dubrava S, Jacobsen J (2014) Phase II crossover trial of varenicline in mild-to-moderate Alzheimer’s disease. Dement Geriatr Cogn Disord 37:232-245.
Lin H, Vicini S, Hsu FC, Doshi S, Takano H, Coulter DA, Lynch DR (2010) Axonal alpha7 nicotinic ACh receptors modulate presynaptic NMDA receptor expression and structural plasticity of glutamatergic presynaptic boutons. Proc Natl Acad Sci U S A 107:16661-16666.
Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK (2012a) Induction of dendritic spines by beta2-containing nicotinic receptors. J Neurosci 32:8391-8400.
Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK (2012b) Glutamatergic synapse formation is promoted by alpha7-containing nicotinic acetylcholine receptors. J Neurosci 32:7651-7661.
McKinney RA (2010) Excitatory amino acid involvement in dendritic spine formation, maintenance and remodelling. J Physiol 588:107-116.
Nordberg A (2001) Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biol Psychiatry 49:200-210.
Nordberg A, Rinne JO, Kadir A, Langstrom B (2010) The use of PET in Alzheimer disease. Nat Rev Neurol 6:78-87.
Oda A, Yamagata K, Nakagomi S, Uejima H, Wiriyasermkul P, Ohgaki R, Nagamori S, Kanai Y, Tanaka H (2014) Nicotine induces dendritic spine remodeling in cultured hippocampal neurons. J Neurochem 128:246-255.
Okada H, Ouchi Y, Ogawa M, Futatsubashi M, Saito Y, Yoshikawa E, Terada T, Oboshi Y, Tsukada H, Ueki T, Watanabe M, Yamashita T, Magata Y (2013) Alterations in alpha4beta2 nicotinic receptors in cognitive decline in Alzheimer’s aetiopathology. Brain 136:3004-3017.
Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N (2004) Cadherin activity is required for activity-induced spine remodeling. J Cell Biol 167:961-972.
Patel SS, Attard A, Jacobsen P, Shergill S (2010) Acetylcholinesterase Inhibitors (AChEI’s) for the treatment of visual hallucinations in schizophrenia: a review of the literature. BMC Psychiatry 10:69.
Placzek AN, Zhang TA, Dani JA (2009) Nicotinic mechanisms in fl uencing synaptic plasticity in the hippocampus. Acta Pharmacol Sin 30:752-760. Radcliffe KA, Fisher JL, Gray R, Dani JA (1999) Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci 868:591-610.
Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT (1998) A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Donepezil Study Group. Neurology 50:136-145.
Rousseau SJ, Jones IW, Pullar IA, Wonnacott S (2005) Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology 49:59-72.
Sabri O, Kendziorra K, Wolf H, Gertz HJ, Brust P (2008) Acetylcholine receptors in dementia and mild cognitive impairment. Eur J Nucl Med Mol Imaging 35 Suppl 1:S30-45.
Scarr E, Gibbons AS, Neo J, Udawela M, Dean B (2013) Cholinergic connectivity: it’s implications for psychiatric disorders. Front Cell Neurosci 7:55.
Tada T, Sheng M (2006) Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol 16:95-101.
Wess J (2004) Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu Rev Pharmacol Toxicol 44:423-450.
10.4103/1673-5374.147943
Hidekazu Tanaka, M.D., Ph.D., Laboratory of Pharmacology, Department of Biomedical Sciences, College of Life Sciences, Ritsumeikan University, hdtanaka@fc.ritsumei.ac.jp.
http://www.nrronline.org/
Accepted: 2014-11-03

- 中国神经再生研究(英文版)的其它文章
- Hydrogen sul fi de controls peripheral nerve degeneration and regeneration: a novel therapeutic strategy for peripheral demyelinating disorders or nerve degenerative diseases
- A novel arti fi cial nerve graft for repairing longdistance sciatic nerve defects: a self-assembling peptide nano fi ber scaffold-containing poly(lactic-co-glycolic acid) conduit
- The effects of claudin 14 during early Wallerian degeneration after sciatic nerve injury
- Transplantation of human amniotic epithelial cells repairs brachial plexus injury: pathological and biomechanical analyses
- Long-term treatment with PP2 after spinal cord injury resulted in functional locomotor recovery and increased spared tissue
- Thermomineral water promotes axonal sprouting but does not reduce glial scar formation in a mouse model of spinal cord injury