茚并异喹啉酮类拓扑异构酶1抑制剂的研究进展*
2014-05-17 09:56:08吴晓明徐进宜
药学与临床研究 2014年3期
丛 蔚,吴晓明,徐进宜
中国药科大学天然药物活性组分与药效国家重点实验室 药学院药物化学教研室,南京 210009
拓扑异构酶 1(Topoisomerase,Top1)是一种广泛存在于生物体(真核细胞、原核细胞)中,参与调节DNA拓扑结构的核酶。DNA在转录和复制的时候,双螺旋打开成为平行的两条单链,两条DNA单链各自双螺旋化和折叠成为两套染色体的过程中Top1都起到决定性作用[1]。因此Top1成为一个良好的药物作用靶点。在正常生理状态下,由Top1导致的DNA断裂是瞬时的,断裂后的再连接速度非常快。通过设计抑制剂插入Top1-DNA断裂复合物(Top1cc)中,使DNA由瞬时断裂变为永久断裂,从而使细胞死亡[2]。
喜树碱是发现的首个Top1抑制剂,通过可逆地与Top1cc形成三元复合物而导致DNA断裂[3]。目前针对喜树碱的改造已经比较全面,很难有太大的突破。而喜树碱很多固有缺陷(来源、与Top1cc结合强度、耐药等)也并没有得到根本解决,因此研发非喜树碱类Top1抑制剂抗肿瘤新药成为共识[4]。经过几十年的努力,科学家研发了众多非喜树碱Top1抑制剂,其中茚并异喹啉酮类最为典型代表。本文对茚并异喹啉酮类的发展历程和改造策略进行详细地综述。
1 茚并异喹啉酮类先导物的发现(1998年)
1978年,Cushman小组[5]在合成氯化两面针碱时,意外地发现一个具有茚并异喹啉酮结构的副产物1(见图1),但当时并没有引起他的重视。直到将近20年之后,Cushman的药理合作者Pommier小组[6]发现1具有较强的抗增殖活性(平均半抑制浓度MGM=8.5 μmol·L-1)和中等的 Top1抑制活性,并将其命名为NSC 314622。
通过充分的药理实验证明,以NSC 314622(1)为代表的茚并异喹啉酮类相对于喜树碱具有以下优点[7]:①通过人工合成得到,相对廉价易得;②化学性质稳定;③插入Top1-DNA复合物的位点与喜树碱不同,因此抗肿瘤谱与喜树碱不同;④与Top1-DNA形成的三元复合物更加稳定,药物作用时间更长;⑤基本不会成为肿瘤过度表达的药物外排泵(ABCG2和MDR-1)的底物,从而有希望抵抗拓扑替康、伊立替康耐药和多药耐药。
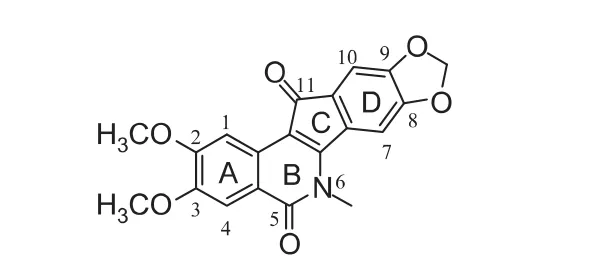
图1 茚并异喹啉酮先导物NSC 314622(1)
NSC 314622(1)是第一个进入临床的茚并异喹啉酮类Top1抑制剂[6]。但遗憾的是,因为其抗肿瘤活性不佳而终止于Ⅰ期临床。1998年以来,Cushman小组针对NSC 314622(1)进行了系统地优化改造,得到了超过400个衍生物,经过药理活性筛选得到许多优良的Top1抑制剂。
2 侧链取代基的初步优化(1998~2006年)
由于茚并异喹啉酮结构新颖,没有可参考的经验,因此只能针对其结构逐一优化探索。先导物NSC 314622(1)由ABCD四个环构成(见图1):A环和D环均为苯环,分别含有2,3-二甲氧基和8,9-亚甲二氧基取代;B环为6元内酰胺环,6位N上有甲基取代;C环是含有酮羰基的五元环。因此初步优化也是针对ABCD环及其取代基开展。同时,Cushman小组针对Top1抑制活性建立了自己的评价体系(见表1)[8]。
2.1 N上取代基的修饰
C环上6位N上取代基是合成过程中最后引入的,因此最容易修饰。将甲基换为更长的疏水(乙基、丁基等)或亲水(羟基、氨基等)侧链发现,N上取代基对于抗增殖和Top1抑制活性至关重要,亲水侧链明显优于疏水侧链(见图2)[8]。改变侧链上氮原子或氧原子分布发现,侧链上氮原子与酰胺氮距离3个碳原子最优[9],如3-氨基丙基或者3-(2-羟乙基胺基)丙基(化合物 4)。 其中后者的 GI50可以达到 0.21 μmol·L-1,与喜树碱类似(++++)[8]。该化合物又被命名为MJ-Ⅲ-65或NSC 706744,并后续开展了详细的临床前实验[10]。进一步将侧链上的直链胺变为环状胺,得到连有咪唑的化合物5(LMP776,Indotecan) 和连有吗啉的化合物 6(LMP400,Indimitecan)[11]。这两个化合物具有良好的抗增殖和Top1抑制活性,而对其他靶点几乎没有作用,因此在2010年进入Ⅰ期临床研究,用于治疗成人复发的各种实体瘤和淋巴瘤[12]。

表1 Top1抑制活性的评价体系
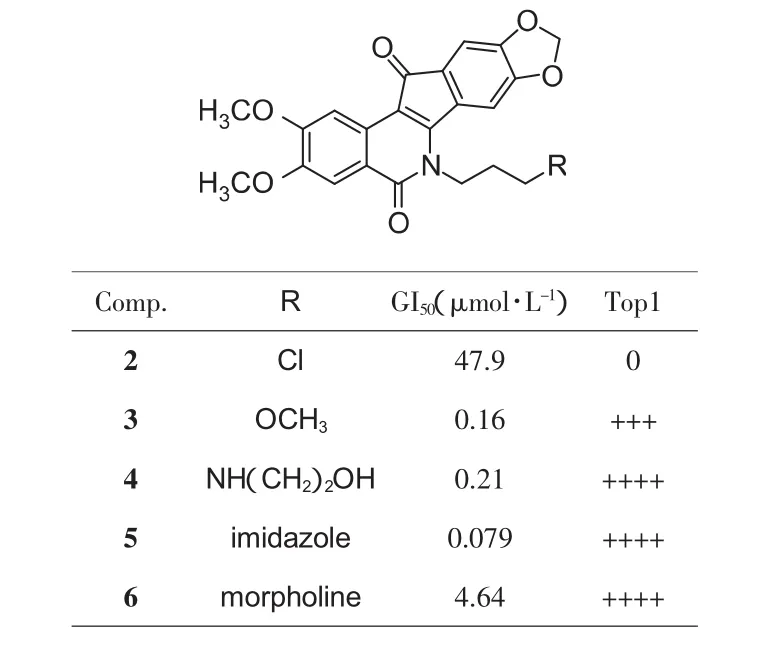
图2 N上取代基的修饰
2.2 A环和D环含氧取代基
最初发现2、3位和8、9位含氧取代可增强抗增殖活性,几个活性很好的化合物均具有含氧取代基(见图2)[9-12]。后续详尽的研究表明[13-14]:①对于活性而言,含氧取代基(尤其是D环)只提供一小部分贡献;②含有甲氧基或者亚甲二氧基取代使药物成为O-脱甲基酶的作用底物,从而易于代谢;③反之,不含氧取代的母核可以提高药物代谢稳定性;④母核上无取代基的茚并异喹啉酮类还有可能作为DNA插入剂而直接作用于DNA,当DNA被其插入后,Top1将无法与DNA结合,也起到抑制DNA功能的作用。
从代谢物中寻找新药是一条捷径,Cushman小组也将这种方法用在茚并异喹啉酮的研究中 (见图3)。2012年,Cushman 小组报道了 LMP776(5)和 LMP400(6)在体内代谢物的研究,发现3-脱甲基得到的3-羟基取代产物(7和8,图3a)活性比原药更好[14]。同样人工合成的其他代谢类似物(9和10,图3b)也显示出优良的活性,提示含氧取代母核在体内可能作用更持久。因此对于AD环的含氧取代基的取舍需要从整体的成药性考虑。
2.3 C环和D环修饰
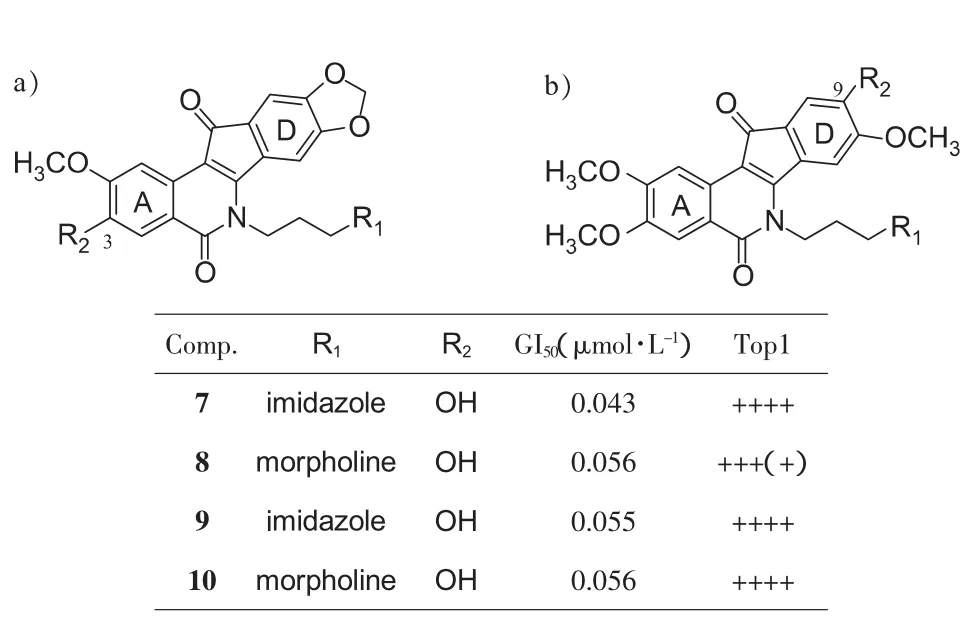
图 3 LMP776(5)和 LMP400(6)体内的脱甲基代谢物
对先导化合物NSC314622(1)进行C环和D环修饰时,发现如下规律:①C环11位酮羰基还原得到11,活性明显下降(见图4a),验证了酮羰基是一个关键的氢键结合位点[9];②将BC环之间的双键还原得到12,抗增殖活性增强,但Top1抑制活性丧失(图4b),可能由于该双键维持骨架平面性起关键作用[15];③将C环酮羰基通过McMurry反应引入11-烯烃长链,末端带有3-氨基取代的化合物得到的13活性最好(图 4c)[16],但并没有超过之前报道的化合物[9,12];④将 D 环由苯环换为萘环得到14,虽然抗增殖活性比先导物强,但是Top1抑制活性明显下降(图4d)[9],可能由于过大的平面体系不利于碱基对的插入以及与Top1的氢键结合。
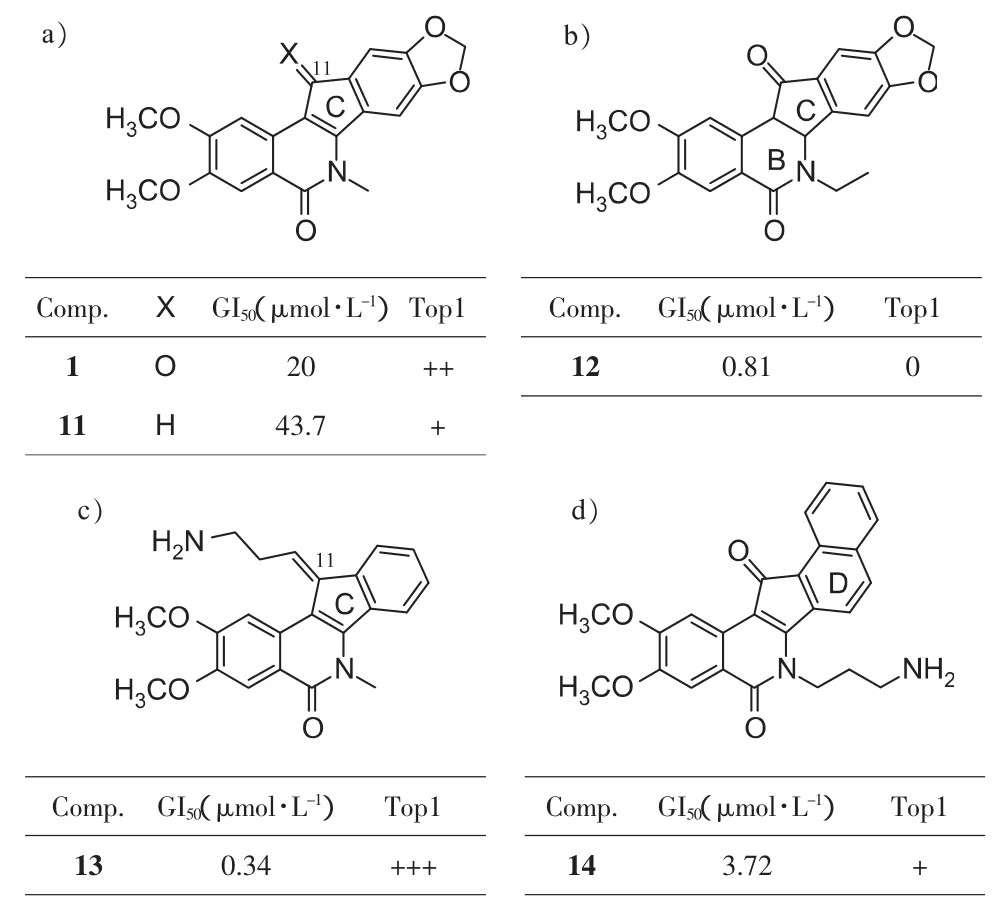
图4 C环和D环的修饰
总之,在最初阶段的修饰中,Cushman小组确证了茚并异喹啉酮母核的关键作用,后续深入的修饰主要是在6位N取代基和AD环上开展。
3 双茚并异喹啉酮类二元插入剂 (2003~2006年)
多胺(Ployamines,PA)是一种广泛存在的胺类,多胺在细胞内质子化后带正电荷,与带负电荷的DNA磷酸基团或者蛋白质作用,从而调控DNA复制、转录和蛋白质的功能[17]。此外,细胞膜上广泛存在多胺转运体系统(polyamine transporter system),因此含有多胺结构的抗肿瘤药物更容易被高多胺需求的肿瘤细胞摄取[17]。
基于上述理论,Cushman小组在母核6位发展了一类长链多胺取代基(图5)。经过研究发现[18]:①第一个氮原子与酰胺氮距离3个碳原子最优;②侧链上氮原子之间间隔2个碳原子最优;③Top1抑制活性并不随氮原子的数目增加而增强,当氮原子数增加到4(17,图5b),Top1抑制活性丧失。经筛选得到的侧链含3个氮原子的化合物15(GI50=2.4 μmol·L-1,++++;图5a)虽然活性与之前报道的最优化合物相似,但是由于侧链上三个铵盐的存在,水溶性大大提高。

图5 多胺侧链
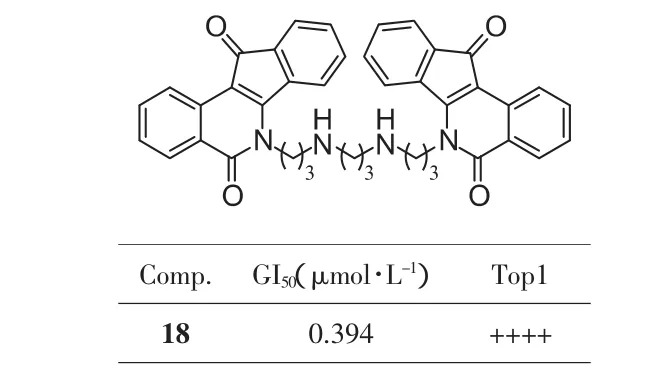
图6 双茚并异喹啉酮类
4 母核电子云分布对于活性的影响(2006~2013年)
2006年[20]至2007年[21-22],Cushman小组继续针对AD环取代基开展研究,发现(图7):将A环供电子的3,4-二甲氧基替换为3-硝基取代后,抗增殖活性显著增强,Top1抑制活性得到保持;而D环只保留9位甲氧基(19)可以使活性得到最优,将其换为氟或溴之后(20和21),抗增殖活性略微降低,替换为体积更大的苯基(22),Top1抑制活性丧失。再对6位侧链的优化同样发现侧链氮原子与母核间隔3个碳原子最好 (23~26)。上述几个最优化合物的活性均超过已发现的所有化合物,而其共有的结构说明3-硝基-9-甲氧基取代至关重要。
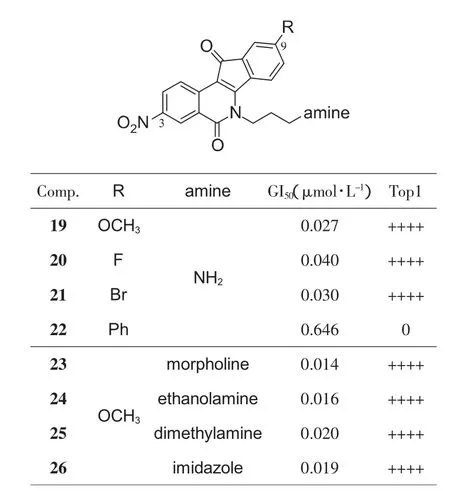
图7 9位取代基的优化
在喜树碱的改造经验中,14位引入氮原子可增加水溶性[23]。2011年以来,Cushman小组也将这个经验应用到茚并异喹啉酮母核的改造上[24-26],在D环上引入氮原子,使水溶性提高一倍至几十倍不等,而活性保持(图8a)[24]。继续比较D环上氮原子位置和甲氧基取代对活性的影响,发现7位氮杂、9位甲氧基取代最佳(图8b)[25]。最终对6位侧链进行优化,侧链末端为咪唑时得到化合物 30(GI50=0.021 μmol·L-1,+++++)[26]。化合物30不仅抗增殖和Top1抑制活性突出,还展示出非常良好的药代动力学特性和抗耐药特性:在针对结肠癌细胞HCT-116的测试中,30与Top1cc可以稳定持续6 h;即使在药物去除后,细胞仍然被阻滞在S期长达3 h;30不是药物外排泵ABCB1的底物;对于几种喜树碱耐药的肿瘤株(KBV.1/Vinbl和 H460/Mito)依然有效[26]。 总之,化合物30是一个非常优良的Top1抑制剂。
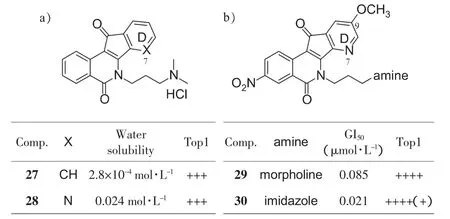
图8 D环和侧链的修饰
从前面对茚并异喹啉酮的修饰过程中可以看出,母核是Top1抑制活性的决定因素。为什么具有3-硝基-7-氮杂-9-甲氧基取代的母核能展示出这么好的活性?Cushman小组发现,除了氢键作用之外,更深层的因素来自于母核的电子云分布[27]。因为母核需要插入Top1cc复合物中,实际是插入相邻两组DNA的碱基对中,所以保持母核平面性是第一个关键因素(反之,将BC环间双键还原导致Top1抑制活性消失[15])。
通过计算机模拟,Cushman小组得出如下结论[27]:①母核与相邻两组碱基对的π-π堆积作用在决定结合取向上起重要影响;②在结合取向确定后,母核与相邻两组碱基对产生静电互补作用,这种静电吸引力对于稳定Top1cc起关键作用;③母核与相邻两组碱基对也可能形成电荷转移相互作用,对于稳定结合力度也起重要作用。所以针对碱基的电子云分布,设计与之电子云互补的母核是提高活性的关键所在。
有了上述的理论支持就很容易解释为什么强吸电的3-硝基和强供电的3-甲氧基取代都对活性有所贡献,但硝基更好[20-22]:3位富电子的基团与邻近缺电子的碱基边缘可以形成静电互补作用;而硝基和甲氧基都是富电子基团,但显然硝基电子云密度更高;此外,硝基的吸电作用使整个母核缺电子,也可以与富电子的碱基母核形成静电互补作用。而D环的7位氮杂、9位甲氧基取代更优的道理与之类似[24-26]。另外,将硝基还原为氨基发现Top1抑制活性丧失,也是因为氨基不具备硝基的特性所导致[20]。
5 糖链的引入(2011年至今)
在另一类非喜树碱Top1抑制剂吲哚咔唑类改造中,吡喃糖侧链是活性必需基团(见图9)[1]。构效关系研究发现,吲哚咔唑类的糖侧链与茚并异喹啉酮6位侧链处于与Top1氢键结合的同一位点[28]。此外,含糖侧链具有很多优点[28-29]:①水溶性好;②糖侧链可与Top1的氨基酸残基形成更强的氢键作用,从而有可能增强活性;③肿瘤细胞膜上过度表达的葡萄糖转运体会使含糖的茚并异喹啉酮更加选择性地被吸收。基于以上优点,Cushman小组采用 “平台跃迁”(platform hop)的策略将吲哚咔唑类的糖链引入到茚并异喹啉酮中。
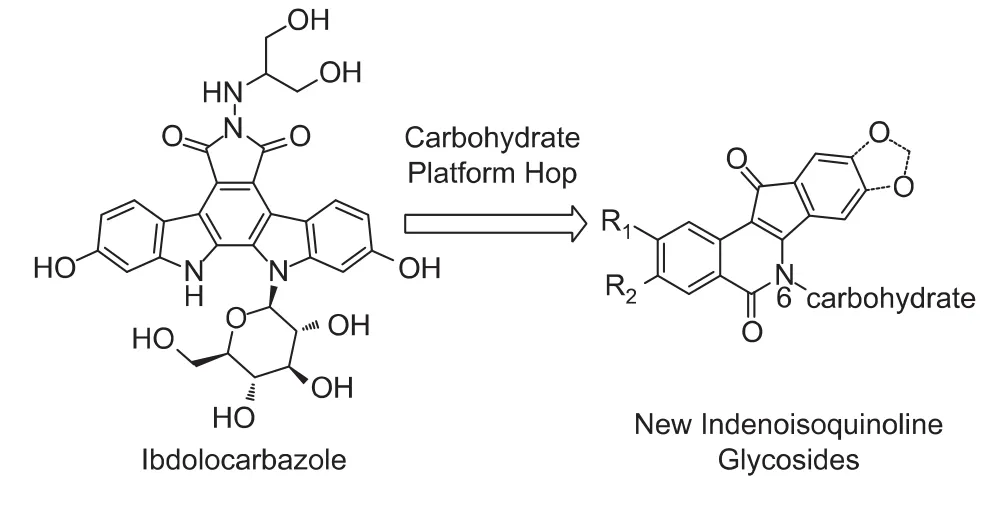
图9 引入糖侧链的设计思想
对含有3、5、6个碳的直链糖、吡喃糖等(图10)化合物的筛选过程中进一步证明,A环3-硝基取代优于含氧取代;C3、C5直链糖的抑制率明显优于 C6(31、32优于33);在吲哚咔唑类中最优的β-吡喃葡萄糖基,在茚并异喹啉酮中却很差(34)[29]。经过筛选得到了十几个活性非常好的化合物(GI50<5 μmol·L-1,Top1 抑制≥++++),初步达到了此次设计的目标。
含糖侧链的茚并异喹啉酮的合成难度明显高于以往的化合物,但它基本指明了后续的研究方向,包括[29]:①在母核上引入之前证明强效的取代基,如3-硝基-7-氮杂-9-甲氧基等;②进一步筛选环状糖和氨基糖等。
6 Top1/Tdp1双重抑制剂(2012年至今)
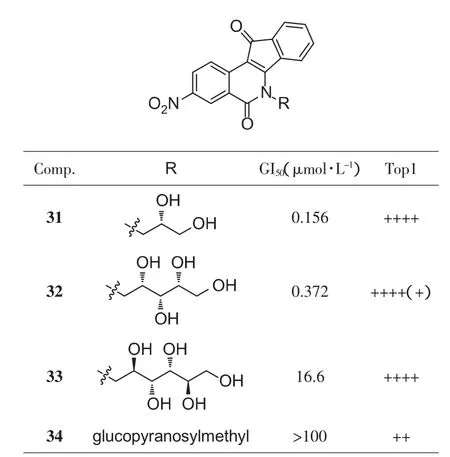
图10 糖链长度优化
Top1与DNA作用形成断裂复合物Top1cc后,自然情况下会瞬时恢复。自然原因或者Top1抑制剂导致的Top1cc阻滞,主要存在两条修复途径。第一条途径通过如下步骤完成[1,3](图11a):①由于DNA被Top1紧紧包裹,泛素-蛋白酶体会先将Top1大部分降解,而只留下与DNA共价结合的部分;②酪氨酰磷酸酯酶 1(Tyrosyl-DNA phosphodiesterase 1,Tdp1)会将Top1残余的酪氨酸-磷酸酯键水解,从而释放出DNA的3′-磷酸末端;③多核苷酸激酶磷酸酯酶(PNK)继续将末端的磷酸去除,从而使3′-羟基游离;④DNA聚合酶和连接酶最后将断开的3′-羟基连接至5′-磷酸,从而完成修复。第二条途径相对简捷(图11b):正常细胞还可以通过检查点激酶(Chk1,2)介导的3′-核苷酸内切酶旁路,将与Top1相连的一段DNA切除,再通过DNA聚合酶和连接酶完成修复[1,3]。
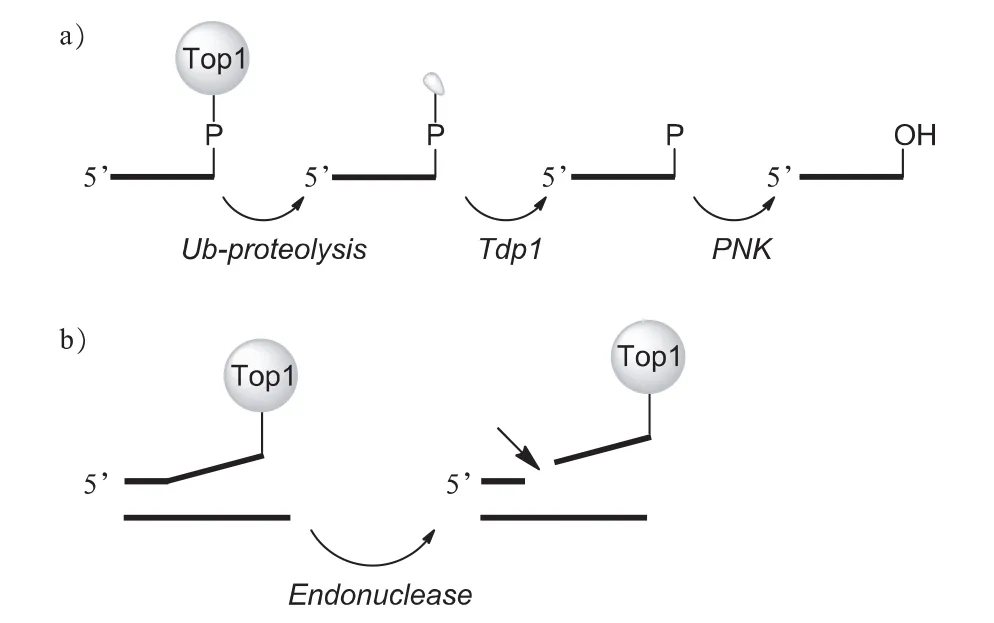
图11 Top1-DNA复合物阻滞修复途径
肿瘤细胞正是由于检查点激酶等抑癌因素缺失才发生癌变,所以第二条途径是肿瘤细胞的先天缺陷(图12b)[30]。当Top1抑制剂导致Top1-DNA复合物阻滞时,肿瘤细胞只能依赖第一条途径完成修复,而其中的Tdp1是完成修复的关键酶,因此设计Tdp1抑制剂可以增强Top1的抑制作用。理论上,Tdp1抑制剂对人体正常细胞毒副作用很小,因为人体细胞可以通过多条途径完成修复(图12a)[30]。因此,Tdp1抑制剂是抗肿瘤的理想靶点。
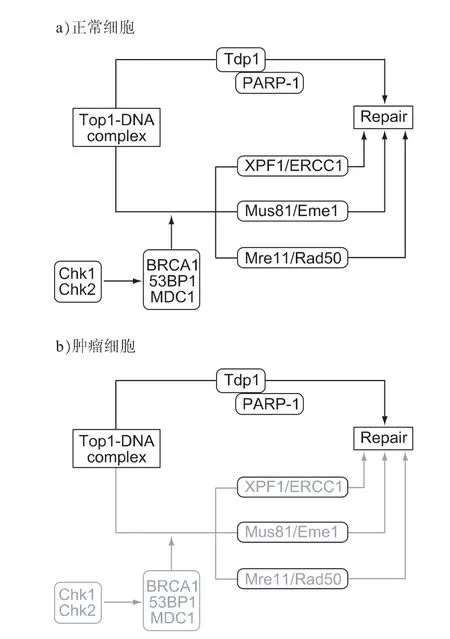
图12 正常细胞与肿瘤细胞修复途径对比
Tdp1的作用底物为磷酸酯键,因此Tdp1抑制剂的设计思路是利用药物模拟磷酸基团的作用,与Tdp1结合而妨碍其催化[30]。Cushman小组基于上述设计思想,开展Top1/Tdp1双重抑制剂的研究。最初,将6位连有长链伯胺的化合物进行了Tdp1抑制活性的筛选(35~37,图13a),尽管它们 Top1抑制活性很差,但其Tdp1抑制活性较好,这表明该类化合物可成为Tdp1抑制剂[31]。文献报道[32]磺酸基团可以模拟磷酸与Tdp1结合,是活性必需基团。因此Cushman小组合成了数十个连有磺酰胺、磺酸酯的化合物。但令人遗憾的是,以化合物40为代表的磺酰胺类,虽然抗增殖和Top1抑制活性很强(GI50=0.046 μmol·L-1,++++;图 13c),但 Tdp1 抑制活性都很差(IC50>111 μmol·L-1)。 反而是未经磺酰化的 6 位末端为伯胺基的化合物(38和39,图 13b)具有中等的 Tdp1抑制活性,Top1抑制活性也比较好(+++)[31]。此外,在筛选之前报道的双茚并异喹啉酮类化合物时意外发现化合物18(图13d)具有很强的 Tdp1 抑制活性(IC50=1.52 μmol·L-1)[31]。 但该化合物可直接作用于DNA,因此Tdp1抑制活性数据可能不准确。综上所述,对于茚并异喹啉酮母核,6位含有3-氨基取代是Tdp1抑制活性的必需基团。
与磺酸类似,羧酸同样可以模拟磷酸基团与Tdp1结合。Cushman小组尝试将羧酸引入茚并异喹啉酮的3位(图14)。化合物 41 虽然具备较强 Tdp1 抑制活性(GI50=5.0 μmol·L-1),但是Top1抑制作用消失,可见同时具备两种抑制活性是非常困难的[32]。Cushman小组又将前述具有6位3-氨基取代的化合物进行筛选,发现了几个同时具备较强Tdp1/Top1抑制的化合物19、42[33]和3-氨基取代的43。
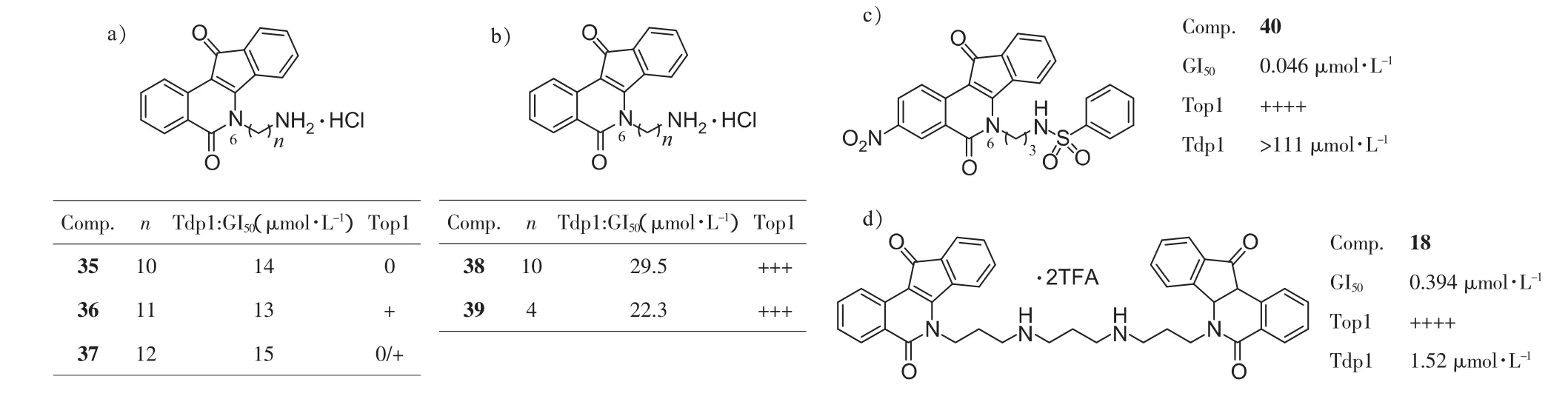
图13 茚并异喹啉酮类Tdp1抑制活性的初步探索
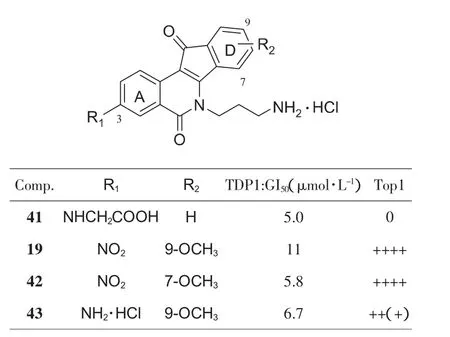
图14 AD环取代基的优化
上述化合物的发现具有重大意义[31,33]:①确证茚并异喹啉酮类可以发展成为Top1/Tdp1双重抑制剂,这也是首次发现的小分子双重抑制剂;②茚并异喹啉酮类母核可以为后续的修饰提供广阔空间,这是其他Tdp1抑制剂所不具备的;③茚并异喹啉酮有可能与Tdp1形成共结晶,从而为基于结构的药物设计提供依据。但是也应该认识到,在上述研究中,Top1、Tdp1和抗增殖活性三者关联并不紧密,因此,还不能得出由于Tdp1被抑制而导致Top1造成的抗增殖活性增强的结论。故研发Top1/Tdp1双重抑制剂还需要继续探索。
7 由茚并异喹啉酮类到吲哚并喹啉类
受Mark Cushman小组研发茚并异喹啉酮类的启发,我们课题组在其基础上,尝试将茚并异喹啉酮类与喜树碱“杂交”(见图15):即保留喜树碱的喹啉环,而将茚并异喹啉酮的异喹啉酮环简化为吲哚环,从而设计了一类与之完全不同而功能相似的吲哚并喹啉母核。吲哚并喹啉类结构稳定,易于合成和修饰,而目前尚无此类母核的Top1抑制活性的报道,具有较强的新颖性。初步的药理活性评价表明,吲哚并喹啉类化合物具有良好的抗增殖活性(GI50<5 μmol·L-1),是非常有希望的抗肿瘤化合物。更加深入的药理活性测试目前正在进行之中。
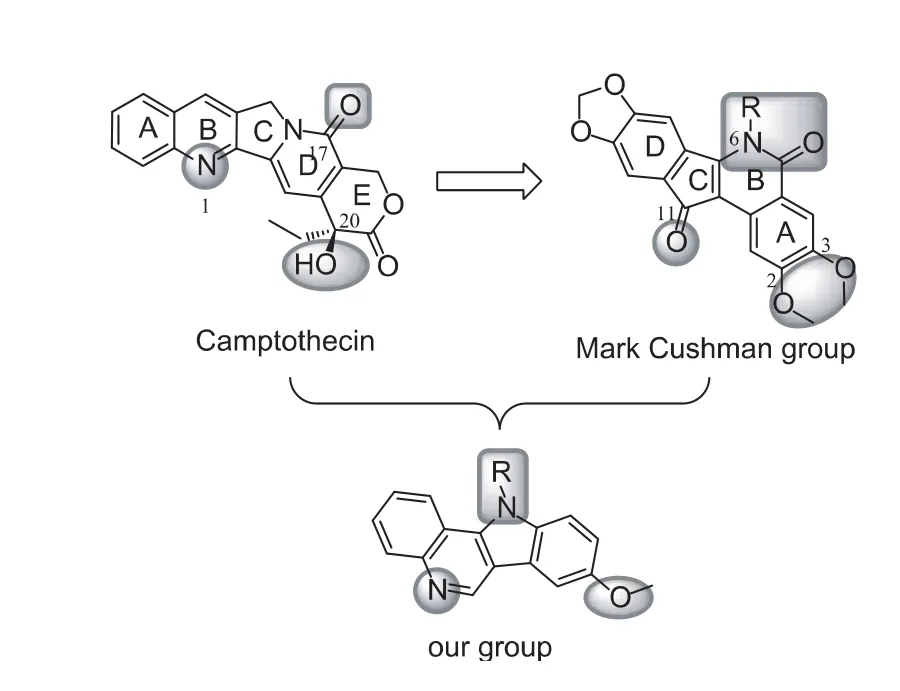
图15 吲哚并喹啉类的设计思想
8 总结与展望
Mark Cushman小组在最近15年的研究中,以NSC 314622(1)为先导化合物,合成了超过400个衍生物,其中有数十个活性优良的Top1抑制剂。最引人注目的是化合物NSC 314622 (1)、Indotecan (5,LMP776) 和 Indimitecan(6,LMP400)先后进入I期临床研究。Cushman小组在对先导化合物优化过程中,综合运用基于结构的药物设计的理念,结合计算机辅助药物设计,加以生物电子等排、前药、代谢物研究等策略,展示了从一个先导化合物到一个茚并异喹啉酮衍生物家族的发展历程。可以预见,未来会有活性更好、成药性更优的茚并异喹啉酮类Top1抑制剂或Top1/Tdp1双重抑制剂被开发出来。茚并异喹啉酮类的发展历程为其他非喜树碱类Top1抑制剂的研究提供了可借鉴的经验。
[1] Pommier Y,Leo E,Zhang H,et al.DNA topoisomerases and their poisoning by anticancer and antibacterial drugs[J].Chem Biol,2010,17(5):421-33.
[2] Pommier Y.DNA topoisomerase I inhibitors:chemistry,biology,and interfacial inhibition[J].Chem Rev,2009,109(7):2894-902.
[3] Wall ME,Wani MC.Camptothecin and taxol:discovery toclinic-thirteenth BruceF.Cain MemorialAward Lecture[J].Cancer Res,1995,55(4):753-60.
[4] Pommier Y.Topoisomerase I inhibitors:camptothecins and beyond[J].Nat Rev Cancer,2006,6(10):789-802.
[5] Cushman M,Cheng L.Total synthesis of nitidine chloride[J].J Org Chem,1978,43(2):286-8.
[6] Kohlhagen G,Paull KD,Cushman M,et al.Proteinlinked DNA strand breaks induced by NSC 314622,a novel noncamptothecin topoisomerase I poison[J].Mol Pharmacol,1998,54(1):50-8.
[7] Pommier Y,Cushman M.The indenoisoquinoline noncamptothecin topoisomerase Iinhibitors:update and perspectives[J].Mol Cancer Ther,2009,8(5):1008-14.
[8] Cushman M,Jayaraman M,Vroman JA,et al.Synthesis of new indeno[1,2-c]isoquinolines:cytotoxic noncamptothecin topoisomerase I inhibitors[J].J Med Chem,2000,43(20):3688-98.
[9] Morrell A,Placzek MS,Steffen JD,et al.Investigation of the lactam side chain length necessary for optimal indenoisoquinoline topoisomerase I inhibition and cytotoxicity in human cancer cell cultures[J].J Med Chem,2007,50(9):2040-8.
[10] Antony S,Kohlhagen G,Agama K,et al.Cellular topoisomerase I inhibition and antiproliferative activity by MJ-III-65(NSC 706744),an indenoisoquinoline topoisomerase I poison[J].Mol Pharmacol,2005,67(2):523-30.
[11] Nagarajan M,Morrell A,Ioanoviciu A,et al.Synthesis and evaluation of indenoisoquinoline topoisomerase I inhibitors substituted with nitrogen heterocycles[J].J Med Chem,2006,49(21):6283-9.
[12] A Phase I Study of Indenoisoquinolines LMP400 and LMP776 in Adults with Relapsed Solid Tumors and Lymphomas.http://clinicaltrials.gov/ct2/show/study/NCT01051635.
[13]Nagarajan M,Morrell A,Fort BC,et al.Synthesis and anticancer activity of simplified indenoisoquinoline topoisomerase I inhibitors lacking substituents on the aromatic rings[J].J Med Chem,2004,47(23):5651-61.
[14] Cinelli MA,Reddy PN,Lv PC,et al.Identification,synthesis,and biological evaluation of metabolites of the experimentalcancer treatmentdrugs indotecan(LMP400)and indimitecan(LMP776)and investigation ofisomerically hydroxylated indenoisoquinoline analogues as topoisomerase I Poisons[J].J Med Chem,2012,55(24):10844-62.
[15] Jayaraman M,Fox BM,Hollingshead M,et al.Synthesis of new dihydroindeno[1,2-c]isoquinoline and indenoisoquinolinium chloride topoisomerase I inhibitors having high in vivo anticancer activity in the hollow fiber animal model[J].J Med Chem,2002,45(1):242-9.
[16] Fox BM,Xiao X,Antony S,et al.Design,synthesis,and biological evaluation of cytotoxic 11-alkenylindenoisoquinoline topoisomerase Iinhibitorsand indenoisoquinoline-camptothecin hybrids[J].J Med Chem,2003,46(15):3275-82.
[17] Battaglia V,Shields CD,Murray-Stewart T,et al.Polyamine catabolism in carcinogenesis:potential targets for chemotherapy and chemoprevention[J].Amino acids,2014,46(3):511-9.
[18] Nagarajan M,Xiao X,Antony S,et al.Design,synthesis,and biological evaluation of indenoisoquinoline topoisomerase Iinhibitors featuring polyamine side chains on the lactam nitrogen[J].J Med Chem,2003,46(26):5712-4.
[19] Nagarajan M,Morrell A,Antony S,et al.Synthesis and biological evaluation of bisindenoisoquinolines as topoisomerase I inhibitors[J].J Med Chem,2006,49(17):5129-40.
[20]Morrell A,Antony S,Kohlhagen G,et al.A systematic study of nitrated indenoisoquinolines reveals a potent topoisomerase I inhibitor[J].J Med Chem,2006,49(26):7740-53.
[21] Morrell A,Placzek M,Parmley S,et al.Optimization of the indenone ring of indenoisoquinoline topoisomerase I inhibitors[J].J Med Chem,2007,50(18):4388-404.
[22] Morrell A,Placzek M,Parmley S,et al.Nitrated indenoisoquinolines as topoisomerase I inhibitors:a systematic study and optimization[J].J Med Chem,2007,50(18):4419-30.
[23] Cheng K,Rahier NJ,Eisenhauer BM,et al.14-Azacamptothecin:a potent water-soluble topoisomerase I poison[J].J Am Chem Soc,2005,127(3):838-9.
[24] Kiselev E,DeGuire S,Morrell A,et al.7-azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents[J].J Med Chem,2011,54(17):6106-16.
[25] Kiselev E,Agama K,Pommier Y,et al.Azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents:A systematic study of structure-activity relationships[J].J Med Chem,2012,55(4):1682-97.
[26]Kiselev E,Sooryakumar D,Agama K,et al.Optimization of the lactam side chain of 7-azaindenoisoquinoline topoisomerase I inhibitors and mechanism of action studies in cancer cells[J].J Med Chem,2014,57(4):1289-98.
[27] Xiao X,Antony S,Pommier Y,et al.On the Binding of Indeno[1,2-c]isoquinolines in the DNA-Topoisomerase I Cleavage Complex[J].J Med Chem,2005,48(9):3231-8.
[28]Peterson KE,Cinelli MA,Morrell AE,et al.Alcohol-,diol-,and carbohydrate-substituted indenoisoquinolines as topoisomerase I inhibitors:investigating the relationships involving stereochemistry,hydrogen bonding,and biological activity[J].J Med Chem,2011,54(14):4937-53.
[29] Beck DE,Agama K,Marchand C,et al.Synthesis and biological evaluation of new carbohydrate-substituted indenoisoquinoline topoisomerase I inhibitors and improved syntheses of the experimental anticancer agents indotecan(LMP400)and indimitecan(LMP776)[J].J Med Chem,2014,57(4):1495-512.
[30] Dexheimer TS,Antony S,Marchand C,et al.Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy[J].Anticancer Agents Med Chem,2008,8(4):381-9.
[31] Nguyen TX,Morrell A,Conda-Sheridan M,et al.Synthesis and biological evaluation of the first dual tyrosyl-DNA phosphodiesterase I(Tdp1)-topoisomerase I(Top1)inhibitors[J].J Med Chem,2012,55(9):4457-78.
[32] Dexheimer TS,Gediya LK,Stephen AG,et al.4-Pregnen-21-ol-3,20-dione-21-(4-bromobenzenesufonate)(NSC 88915)and related novel steroid derivativesastyrosyl-DNA phosphodiesterase (Tdp1)Inhibitors[J].J Med Chem,2009,52(22):7122-31.
[33] Conda-Sheridan M,Reddy PN,Morrell A,et al.Synthesis and biological evaluation of indenoisoquinolines thatinhibitboth tyrosyl-DNA phosphodiesterase I(Tdp1)and topoisomerase I(Top1)[J].J Med Chem,2012,56(1):182-200.
猜你喜欢
山东农业大学学报(自然科学版)(2021年3期)2021-07-29 03:07:02
新生代(2019年8期)2019-10-28 06:39:30
世界农药(2019年2期)2019-07-13 05:55:12
天然产物研究与开发(2018年7期)2018-08-21 02:04:02
中成药(2018年2期)2018-05-09 07:20:05
天然产物研究与开发(2018年4期)2018-05-07 06:47:52
粘接(2017年4期)2017-04-25 08:37:20
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
中国塑料(2015年10期)2015-10-14 01:13:13