
全基因组测序及其在遗传性疾病研究及诊断中的应用
2014-05-10 01:25邵谦之姜毅吴金雨
遗传 2014年11期
邵谦之,姜毅,吴金雨
1. 温州医科大学基因组医学研究院,温州 325000;
2. 中国科学院北京生命科学研究院,北京 100101
随着高通量测序技术(Next generation sequencing,NGS)的不断发展,特别是随着测序费用的逐年降低以及数据分析流程的日趋成熟,全基因组测序(WGS)已经成为疾病研究、临床诊断中重要的手段[1,2]。研究者已经运用全基因组测序来检测癌症、孟德尔遗传病、复杂疾病的致病突变和致病基因,取得了前所未有的科研成果[3]。本文就全基因组测序的数据分析及其在疾病研究和临床诊断中的应用进行综述。
1 全基因组测序的背景介绍
近年来,随着高通量测序技术的不断发展与成熟,全基因组测序被应用到了各种领域,尤其是在遗传性疾病研究方面的应用备受关注[1,2,4~6]。目前人类已知的疾病中,大约有4000多种疾病与基因异常有关[7]。利用全基因组测序,可在全基因组水平上检测与人类疾病相关的单核苷酸变异(SNVs)、插入缺失(InDels)、拷贝数变异(CNV)和结构变异(SV)等多种全面的突变信息,进而找到致病突变并研发有效的治疗药物,为临床用药提供指导。
价格昂贵一直是全基因组测序发展的一个重大阻碍,然而随着Hiseq X Ten的出现,全基因组测序的成本已大幅下降,测序费用仅需1000美元。Hiseq X Ten是由Illumina公司研发的有史以来最强大的测序平台,旨在提供大规模人类基因组测序服务。它由10台超高通量测序仪组成,每台测序仪的产出效率是Hiseq 2000的12倍,每天可产出高达600 GB的数据量,全年可以完成约18000人次全基因组测序。数据分析速度慢则是全基因组发展的另一个难题,受数据量及分析软件的限制,全基因组数据分析需要1 d以上。然而2014年7月,Dutch生物信息公司宣布开发的Genalice Map软件可以成功实现1 min比对人类全基因组,并在将来的合作中继续测试10000个人类全基因组。此外,由Edico Genome开发的生物科技处理器(Dynamic Read Analysis for Genomics,DRAGEN),作为全球首款新一代测序生物信息特殊应用集成电路,可以将用于分析整个人类基因组数据所需的24 h锐减为18 min,同时还确保了分析的准确性。相信不久以后,其他分析步骤也将在几分钟内完成。
尽管全基因组测序面临着价格昂贵、数据分析速度慢等难题,但是由于其能检测结构变异以及非编码区的SNVs、InDels等,目前在国内已被应用于一系列遗传性疾病的研究。早在2003年,赵国屏课题组就利用全基因组测序分析钩端螺旋体病[8]。此后,全基因组测序逐渐被应用于肝癌[9]、膀胱癌[10]、胰腺癌[11]、腹膜间皮瘤[12]、自闭症[13]等疾病致病机理的研究。谢晓亮课题组于2012年底利用其新近发明的MALBAC扩增技术对一个亚洲男子的99个精子进行单细胞全基因组DNA扩增,首次实现了单个精子高覆盖度的全基因组测序[14]。此外,该课题组还首次利用上述 MALBAC基因组扩增高通量测序对试管婴儿进行单基因遗传病筛查,该婴儿已于 2014年9月19日在北京大学第三医院诞生,标志着我国胚胎植入前遗传诊断技术已处于世界领先水平。由此可见,全基因组测序已成为现阶段基因测序工作的重心。全基因组测序的时代已经到来,势不可挡。
2 全基因组测序的数据分析流程
全基因组测序的数据分析流程包括质量控制(Quality control)、比对(Mapping)、突变检测(Call variant )、突变注释(Annotation)。针对不同数据要求,已有多款分析软件得以开发(表1),目前广泛使用的分析流程为“BWA+ GATK + ANNOVAR”(附图 1)。
2.1 质量控制
对测序产生的原始数据(Raw data)进行去接头、过滤低质量处理,得到 Clean data的过程称为质量控制。质量控制能除去部分测序效果较差的序列,提高后续分析的准确性。经过该步骤通常会过滤掉5%~15%低质量的序列。
2.2 比对到参考基因组
将质量控制后的Clean data比对到参考基因组上,得到每条序列的比对位置、比对质量值等信息。目前最主流的比对软件为 BWA(Burrows-Wheeler Aligner)[18],它能将短序列准确快速地比对到参考基因组上,生成通用的 SAM 格式的文件。自 2013年起 BWA发布了新算法 BWA MEM,可以比对70bp~1 Mb的序列,比原来的算法更加准确,运行速度也更加快[54]。
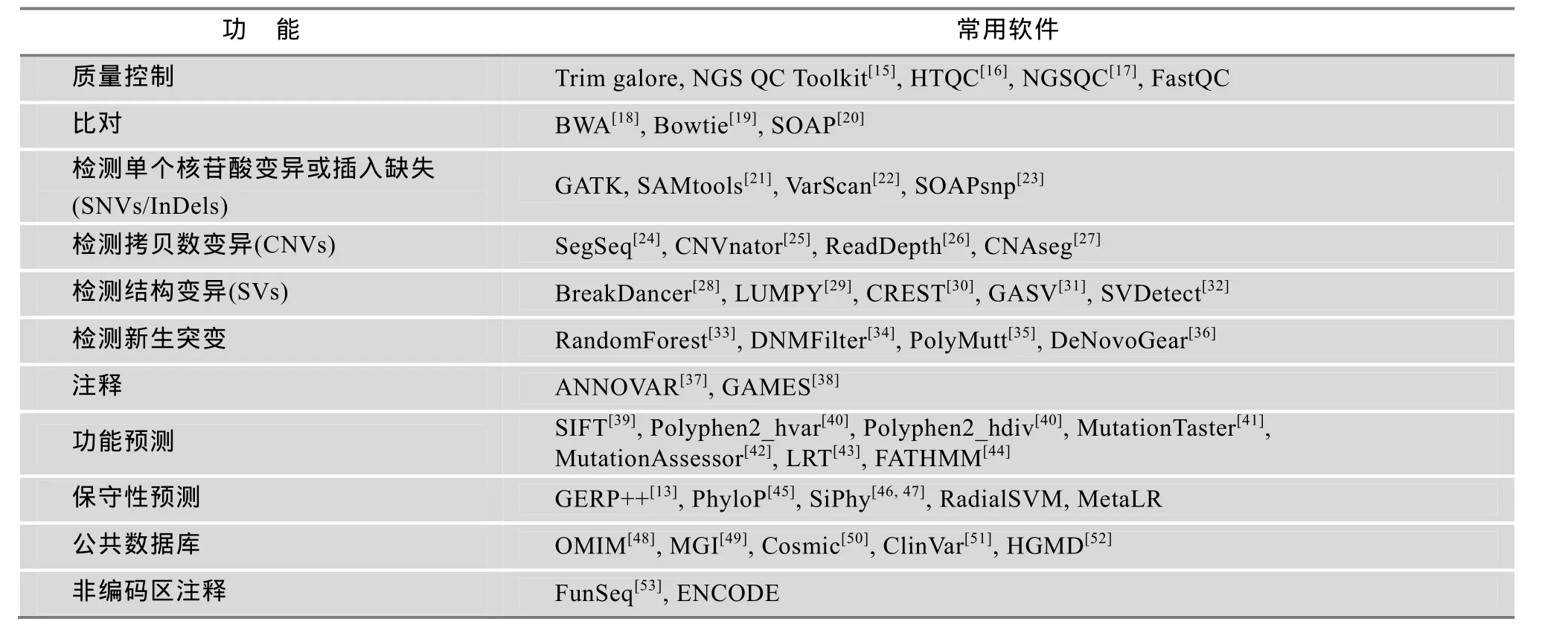
表1 全基因组数据分析常用软件
2.3 突变检测
比对好的SAM文件通常会转换成BAM文件并进行去重(Remove duplication),然后进行突变的检测。目前主流检测SNV和InDel的软件为Genome Analysis Toolkit (GATK,http://www.broadinstitute.org/gatk/),GATK准确度非常高,它会对BAM文件进行两次校正过程以提高突变检测的准确率,但是速度比较慢。2014年 3月,Broad宣布最新版GATK(version3.1)在突变检测速度上将比原来快3~5倍,使全基因组的分析时间从3 d缩短到1 d。
由于全基因组测序具有较好的均一性和覆盖度,因此在 CNVs的检测方面具有众多优势。目前已经发表了多种CNV的检测方法与软件,可以分为两大类别:(1)基于深度差异的检测方法受测序局部不均一性的影响,往往假阳性率比较高; (2)基于读段对之间的距离检测 CNV的方法能相对准确地找到断点。若读段对之间的距离明显超过正常大小,就可以认为这对读段之间存在 CNV。另外,有些比对不上的读段拆成两条读段后能分别比对到染色体上不同位置,这两个位置之间也可能存在 CNV。广义上的SVs包括CNVs和倒位、易位等多种类别,因此SVs的检测比 CNVs更为复杂,往往需要多款软件结合使用,才能更准确地找到可能的SVs。CNVs和SVs都需要通过Sanger测序对断点进行验证才能最终确定,如果无法确定断点的则需要通过 qPCR验证。
最近,越来越多研究表明新生突变(de novo mutation)在散发性疾病中扮演重要的角色[55],特别是在神经精神疾病中鉴定到一系列的致病基因[56,57]。因此,具有核心家系(例如:患者以及患者的父亲与母亲)的全基因组测序也开始得到广泛应用。目前已经开发出了一系列的软件与工具,这些软件对多个样品同时鉴定突变,并筛选出仅在患者出现突变。新生突变通常都是极端稀有,对散发性疾病具有重要作用。
2.4 注释突变及预测致病基因
每一个全基因组的样品,平均可以检测到大约3000000个突变。为了筛选致病的候选突变并用于后续功能验证,需要通过诸如 ANNOVAR[37]等软件对其进行注释。一方面,利用已知突变数据库(如dbSNP139[58]、ESP6500[59]、1000 Genome[60]等),去除在数据库中出现频率较高的突变,并将剩下的突变注释到基因组上的各个基因区间(如外显子区、内含子区、5′-UTR区或3′-UTR区)和突变对蛋白质编码的改变情况(如错义突变、无义突变或移码突变);另一方面,通过多个疾病数据库(OMIM[48]、MGI[49]、Cosmic[50]、ClinVar[51]、HGMD[52]等)将部分已知突变与疾病表型联系起来,并利用多款预测软件(如SIFT[61]、Polyphen[62]、GERP++[63]、LRT[64]等)对这些突变进行有害性和保守型预测,最终鉴定导致疾病发生的相关基因及突变。
随着科研人员对遗传性疾病的进一步研究,发现在非编码区域,特别是一些位于高度保守区域、启动子区域以及重要调控区域的突变对疾病的发生仍然具有不可替代的作用[65,66]。非编码区的功能分析常用 FunSeq[53]软件进行。FunSeq过滤掉 1000 genomes中的突变后,根据突变是否在某些功能元件上、是否在敏感区域、是否中断转录因子模体、靶标基因是否已知及靶标基因是否在网络中心等对剩下的突变进行打分,筛选出可能有害的突变。如果有多个样本一起分析,FunSeq还可以判断一个突变是否是频发突变(Recurrent mutation)。另外,还需要充分利用ENCODE数据库(http://genome.ucsc.edu/ENCODE/),里面包含了多种细胞系不同功能元件的注释信息(如启动子、增强子、转录因子等),可以为非编码的研究提供参考。
3 全基因组测序在疾病研究及临床诊断中的应用
全基因组测序给疾病研究以及致病基因的筛选带来了前所未有的机遇。近年来,通过全基因组测序方法,已在孟德尔遗传病、癌症等疾病中鉴定到了一系列的致病突变和基因,已经成为致病基因鉴定和临床诊断的重要手段之一[4,7]。
3.1 在癌症中的应用
癌症是指细胞的生长与增生不受机体控制,从而引起机体功能受损的一类疾病。癌细胞的基因组缺乏稳定性,容易发生各种突变,进而改变细胞功能,使患者产生一系列的临床症状。高通量测序技术特别是全基因组测序对癌症中体细胞突变的鉴定,疾病的诊断与治疗提供了最直接有效的方法之一,并得到了广泛应用。通过全基因组测序,许多癌症已经被广泛研究,并取得了一系列的研究成果。Pleasance等[67]在 2010年首次通过全基因组测序得到了黑色素瘤的全基因组突变谱。他们发现,黑色素瘤的体细胞突变在基因组上面不均一分布,绝大部分的突变都是C>T/G>A这种类型,而这些突变绝大部分发生在CpC/GpG上面。产生这种特异突变普的原因可能是黑色素瘤患者长期暴露于紫外线照射中。 Pleasance等[68]采用全基因组测序技术,在小细胞肺癌中却发现 G>T/C>A转换在所有突变中占主要部分,并且更倾向于发生在 CpG上面,揭示这种特殊的突变谱可能与患者的长期吸烟有关。Lee等[69]对肺癌进行全基因组测序却发现 C>T/G>A转换占突变的比例最高,并且富集于 CpG上面,暗示可能同甲基化的脱氨基作用有关。由于不同癌症具有不同的发病机理,因此可能会表现出不同的突变谱。全基因组测序提供了最直接有效、无偏向性地的方法系统分析癌症突变谱,为深入了解致病机理提供指导。
不同癌症不但具有特异的突变谱,同时还具有不同的突变频率,差距可能达到1000倍以上[70]。横纹肌样瘤的突变频率最小,每一Mb区域约发生0.1个突变; 然而黑色素瘤的突变频率最高,达到100/Mb。研究表明,组织差异性可能是造成突变频率差异最直接的原因,而且受较大外界压力(如吸烟、紫外线照射等)的癌症通常具有较高的突变频率。另外,同一种癌症的不同患者携带的突变数量同样具有很大的差异性。例如,在黑色素瘤和肺癌中,突变频率最少的样品只有 0.1/Mb,而突变频率最高的样品却达到100/Mb以上。尽管如此,研究者们使用全基因组技术,从 SNVs、InDels、CNVs和 SVs等多个角度寻找致病突变,找到一系列可复制的致病基因。Puente等[71]对 4对慢性淋巴细胞性白血病(Chronic lymphocytic leukaemia)样品进行全基因组测序,鉴定到46个对蛋白功能有害的突变。大样本量验证后发现 4个基因(NOTCH1、XPO1、MYD88和KLHL6)携带复发突变。Roberts等[72]对15例急性淋巴细胞白血病样品进行全基因组测序,在多个基因(ABL1、JAK2、PDGFRB、CRLF2 和 EPOR)中发现了结构变异,同时在 IL7R、FLT3和 SH2B3基因中鉴定到多个害突变。对这些基因的功能进行深入分析后发现,体细胞突变减弱了相应蛋白同络氨酸激酶抑制剂的结合,因此与络氨酸酶抑制剂相关的药物对这些患者的定向治疗将具有重要临床指导意义。最近,Wang等[73]使用全基因组测序技术,对100对胃癌样品进行全面分析,包括编码区域和非编码区域的点突变、插入缺失、拷贝数变异、结构变异、基因表达以及甲基化图谱,成功鉴定已知的胃癌致病基因(TP53、ARID1A 和 CDH1)以及新的胃癌致病基因(MUC6、 CTNNA2、GLI3和RNF43等)。通过全基因组测序,已经在白血病[71,72,74]、黑色素瘤[75]、脑膜瘤[76]、乳腺癌[77]、成神经管细胞瘤[78]、肾癌[79]、小细胞肺癌[80]、结肠癌[81]和甲状腺癌[82]等多种癌症中鉴定到一系列的致病突变和基因。
由于全基因组测序对结构变异与非编码区变异的检测具有无可比拟的优势,该技术已经全面应用于癌症领域,使得科研工作者对癌症的发生发展有更深入的了解。随着测序成本的降低以及数据分析手段的发展,更多的癌症和样品将被测序,并鉴定到一系列有可重复的致病基因。为了更好的研究癌症,科研工作者们已经成立了国际基因组联盟(International Cancer Genome Consortium,ICGC),到目前为止该联盟已经公布了超过10000个癌症基因组数据。全基因组测序已经成为癌症研究的工作重心,有益于系统分析致病基因参与的分子通路,将为临床用药提供最有效依据,使得癌症的治愈也将成为可能。
3.2 在神经与精神疾病中的应用
全基因组测序技术不仅在癌症等疾病中得以应用,也逐步被应用到其他常见遗传病中,尤其是神经与精神疾病。全基因组测序在结构变异的鉴定方面存在无可比拟的优势,可以准确的找到断点位置,精确定位致病基因。Talkowski等[83]对具有神经发育障碍的患者进行全基因组测序并鉴定到 33个区域。这些区域的致病基因可以归类为4种类别:(1)已知的致病基因(AUTS2、FOXP1和CDKL5); (2)单个基因的区域(SATB2、EHMT1); (3)新的候选基因与区域(CHD8、KIRREL3和ZNF507); (4)同其他神经精神疾病相关的基因(TCF4、ZNF804A、PDE10A、GRIN2B和ANK3)。他们的研究表明多个基因可能共同作用,并产生多种多样的表型。Michaelson等[33]在2012年对10个自闭症谱系障碍(Autism spectrum disorder,ASD)核心家系(患者以及正常的父母亲)进行全基因组测序。分析发现新生突变在全基因组范围内的分布不是随机的,而是存在一定的热点区域,而且这些热点区域同疾病具有重要的关系。他们还发现基因组不同区域的突变速速率同基因组中的多种因素(如 CG含量、复制时间、转录水平和敏感位点等)存在一定联系。最终他们提出了一种回归模型,引入上面多种因素,可以准确地计算自闭症患者在基因组不同区域的突变速率,为热点区域的鉴定提供参考和依据。同时他们还发现公共数据库中的致病基因,不管是显性遗传还是隐性遗传都具有较高的突变速率。此外, Kong等[84]在全基因组水平证明新生突变的个数与父亲的年龄存在着显著的关系,而不是母亲的年龄。而且父亲的年龄每增加一岁,小孩携带的平均新生突变个数将增加两个,从而增加了患神经精神疾病的风险。
科研人员通过全基因组测序不但揭示了突变发生的一些本质规律,同时还有效地鉴定了一系列致病基因。Jiang等[13]对32个自闭症(ASD)核心家系进行全基因组测序,最大可能地将临床表型同遗传变异联系起来,从新生突变、稀有遗传变异等多个角度进一步解释ASD的发病机理。他们的研究鉴定到一系列与 ASD 相关的致病基因,包括 CAPRIN1、AFF2、VIP、SCN2A、KCNQ2和 CHD7等。针对ASD这类具有高度异质性的遗传病,全基因组测序能够更有效地鉴定致病突变与基因。最近,Nature杂志发表了对 50个智力残疾的核心家系进行全基因组测序的研究,鉴定到84个在编码区域的新生突变,以及8个新生CNVs[5]。得力于与全基因测序的高覆盖度和均一性,能够对 62%的患者进行临床诊断,找到明确的致病基因,充分肯定了全基因组测序的重要意义。
全基因组测序在神经精神疾病的运用才刚刚开始,更多的基因组测序将被完成。例如,中美科研机构将合作完成“万人自闭症基因组研究计划”。 这个项目有助于更全面地了解、发现绝大多数自闭症儿童患病原因,并能应用于对自闭症儿童的早期临床诊断和家庭的产前筛查,最终了解自闭症的发病机理并开发出有效治疗方法。总之,全基因组测序将在神经精神疾病中得到更为广泛的应用。
3.3 在临床诊断中的应用
全基因组测序技术不仅在疾病致病基因的研究中扮演着重要的角色,它还广泛地应用于临床上一些疾病的诊断、筛查,为疾病的预防以及治疗提供依据。目前,大多数的产前诊断都是基于有创性的侵入检查手段,如羊膜腔穿刺术、胎儿脐带血穿刺等。这种侵入性技术对孕妇以及婴儿都存在一定的伤害,甚至可能导致流产[85,86]。侵入性产前诊断通过分析母亲血样中的胎儿DNA,避免了穿刺损失、感染和流产的风险,减轻了孕妇的精神压力,易为广大孕妇和家属接受。 目前,全基因组测序已在无创产前诊断(Non-invasive prenatal testing,NIPT)领域显现雏形。一方面,可通过全基因组测序技术,非侵入性检查染色体非整倍异常,为 21三体综合征、18三体综合征等的准确诊断提供了一个有效的解决方案。Lau等[87]通过全基因组测序技术对 5例孕妇进行无创产前诊断,并在 4例中发现了染色体异常,准确诊断出21三体综合征、18三体综合征等。Lau等[2]通过全基因组测序对 1982例样品的游离 DNA进行分析,并证明即使深度较低(0.1x),全基因组测序也能检测到染色体结构变异,对常见的三体综合征做出准确的诊断。另一方面,还可通过全基因组测序,非侵入性诊断诸如癌症等基因异常性疾病。Leary等[88]对 10例结直肠癌、乳腺癌样品以及 10例正常人进行全基因组测序,在 ERBB2基因和CDK6基因找到了染色体拷贝数变异和重排,并证明可以不依赖活组织检查而进行无创诊断。
随着高通量测序技术的发展,以高通量、自动化、高准确度为显著特征的第二代测序技术(NGS)已被成熟地运用于一些疾病的诊断和筛查。其中,基于全外显子测序技术的基因诊断已成功地对先天性氯腹泻[89]、新生儿糖尿病[90]、难治性炎性肠病[91]和 Charcot-Marie-Tooth atrophy综合征[92]等疾病进行分子水平的诊断。但是由于该技术对结构变异与非编码区变异的研究具有局限性,在一些由结构变异或非编码区变异所导致的复杂疾病(如 21三体综合征)面前则力不能及。
4 全基因组测序数据分析面临的挑战
尽管全基因组测序能够有效地挖掘全基因组范围内的多种变异,为遗传性疾病的研究以及临床诊断提供极大便利。但是,由于下机数据量的巨大增加给全基因组测序的数据分析带来巨大挑战。(1)数据存储:一个标准的全基因数据通常在100 GB左右,再加上分析得到的clean data、BAM文件、SAM文件以及突变结果文件,一个全基因组数据往往还需要额外300 GB的存储空间。例如,100个标本的全基因组数据,完成所有数据分析至少需要 30 TB以上的存储空间。(2)数据分析效率:如此巨大的数据将给数据分析效率以及服务器的运算性能带来巨大的挑战。数据分析过程中往往需要使用多线程,同时还需要将数据分成多份同时运算,以加快数据分析效率。(3)筛选致病变异:通常情况下,通过全基因组测序将分析得到大约 3000000个 SNV以及InDels,如何从如此众多的突变中,特别是非编码区域重要调控原件中寻找致病突变成为亟待解决的问题。与此同时,还有可能找到多个CNV/SV,如何确定这些变异对疾病的贡献也存在巨大挑战。(4)CNV/SV鉴定的准确率:尽管目前发表了多款基于全基因组测序鉴定 CNV/SV的方法与工具,但是准确率都不高,同时还存在一定的假阴性。尽管如此,在阐明疾病的发病机理时,全基因组测序在疾病的基因诊断和致病基因的研究中仍具有不可替代的作用。
5 展 望
目前,全基因组测序技术已在疾病研究和临床诊断中得到日益广泛的应用,特别是对妊娠过程中母体血浆中存在游离的胎儿DNA (Fetal DNA)[93]通过全基因组测序进行无创产前诊断。另一方面,随着大数据时代的来临,为了使大数据能够得到更快的分析和更有效的利用,全基因组测序必然向着数据的云存储、云计算等方向发展。(1)云存储:基于分布式原理存储高通量数据,极大地降低数据分析时输入、输出和中间数据量,从而加快数据分析速度,在相同的时间里处理更多的测序数据; (2)云计算:开发基于并行原理的生物信息学软件,并行地处理高通量数据,提高数据分析过程中每个步骤的效率,充分利用计算资源,从而消耗资源更少,数据分析更迅速; (3)测序与分析一体化:即把高通量测序与后续数据分析相结合,下机得到的数据不仅有测序结果,还有检测到的各种突变(SNVs、InDels、CNVs或 SVs),并与云存储的疾病数据库相关联,以预估病人各种遗传疾病的风险。目前已有相关的工具得以开发如 MegaSeq[94],利用全基因组测序进行疾病的基因诊断和致病基因的研究将是一个非常有前景的领域。又如,biobambam[95]可以在不损害比对重要信息的情况下,对 BAM 文件进行大幅度压缩100倍以上,极大地缩减了数据存储空间。最近,基因组学重要软件 SAMtools也在基因组数据量的快速上升的背景下进行了重要升级,最新版本支持压缩和全球共享数据。总而言之,全基因组测序的时代已经到来,将会在遗传性疾病的研究和临床诊断中发挥更重要的作用。
附录
附图1见文章电子版(www.Chinagene.cn)。
[1]Dewey FE,Grove ME,Pan CP,Goldstein BA,Bernstein JA,Chaib H,Merker JD,Goldfeder RL,Enns GM,David SP,Pakdaman N,Ormond KE,Caleshu C,Kingham K,Klein TE,Whirl-Carrillo M,Sakamoto K,Wheeler MT,Butte AJ,Ford JM,Boxer L,Ioannidis JP,Yeung AC,Altman RB,Assimes TL,Snyder M,Ashley EA,Quertermous T. Clinical interpretation and implications of whole-genome sequencing. JAMA,2014,311(10): 1035–1045.
[2]Lau TK,Cheung SW,Lo PSS,Pursley AN,Chan MK,Jiang F,Zhang H,Wang W,Jong LFJ,Yuen OKC,Chan HYC,Chan WSK,Choy KW. Non-invasive prenatal testing for fetal chromosomal abnormalities by low-coverage whole-genome sequencing of maternal plasma DNA: review of 1982 consecutive cases in a single center. Ultrasound Obst Gyn,2014,43(3): 254–264.
[3]Cirulli ET,Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet,2010,11(6): 415–425.
[4]Rabbani B,Tekin M,Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet,2014,59(1): 5–15.
[5]Gilissen C,Hehir-Kwa JY,Thung DT,van de Vorst M,van Bon BWM,Willemsen MH,Kwint M,Janssen IM,Hoischen A,Schenck A,Leach R,Klein R,Tearle R,Bo T,Pfundt R,Yntema HG,de Vries BBA,Kleefstra T,Brunner HG,Vissers LELM,Veltman JA. Genome sequencing identifies major causes of severe intellectual disability. Nature,2014,511(7509): 344–347.
[6]Egan JB,Shi CX,Tembe W,Christoforides A,Kurdoglu A,Sinari S,Middha S,Asmann Y,Schmidt J,Braggio E,Keats JJ,Fonseca R,Bergsagel PL,Craig DW,Carpten JD,Stewart AK. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events,evolution,and clonal tides.Blood,2012,120(5): 1060–1066.
[7]Boycott KM,Vanstone MR,Bulman DE,MacKenzie AE.Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet,2013,14(10): 681–691.
[8]Ren SX,Fu G,Jiang XG,Zeng R,Miao YG,Xu H,Zhang YX,Xiong H,Lu G,Lu LF,Jiang HQ,Jia J,Tu YF,Jiang JX,Gu WY,Zhang YQ,Cai Z,Sheng HH,Yin HF,Zhang Y,Zhu GF,Wan M,Huang HL,Qian Z,Wang SY,Ma W,Yao ZJ,Shen Y,Qiang BQ,Xia QC,Guo XK,Danchin A,Girons IS,Somerville RL,Wen YM,Shi MH,Chen Z,Xu JG,Zhao GP. Unique physiological and pathogenic features of Leptospira interrogans revealed by wholegenome sequencing. Nature,2003,422(6934): 888–893.
[9]Kan ZY,Zheng HC,Liu X,Li SY,Barber TD,Gong ZL,Gao H,Hao K,Willard MD,Xu JC,Hauptschein R,Rejto PA,Fernandez J,Wang G,Zhang QH,Wang B,Chen RH,Wang J,Lee NP,Zhou W,Lin Z,Peng ZY,Yi K,Chen SP,Li L,Fan XM,Yang J,Ye R,Ju J,Wang K,Estrella H,Deng SB,Wei P,Qiu M,Wulur IH,Liu JG,Ehsani ME,Zhang CS,Loboda A,Sung WK,Aggarwal A,Poon RT,Fan ST,Wang J,Hardwick J,Reinhard C,Dai H,Li YR,Luk JM,Mao M. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res,2013,23(9): 1422–1433.
[10]Guo GW,Sun XJ,Chen C,Wu S,Huang PD,Li ZS,Dean M,Huang Y,Jia WL,Zhou Q,Tang AF,Yang ZQ,Li XX,Song PF,Zhao XK,Ye R,Zhang SQ,Lin Z,Qi MF,Wan SQ,Xie LF,Fan F,Nickerson ML,Zou XJ,Hu XD,Xing L,Lv ZJ,Mei HB,Gao SJ,Liang CZ,Gao ZB,Lu JX,Yu Y,Liu CX,Li L,Fang XD,Jiang ZM,Yang J,Li CL,Zhao X,Chen J,Zhang F,Lai YQ,Lin ZG,Zhou FJ,Chen H,Chan HC,Tsang S,Theodorescu D,Li YR,Zhang XQ,Wang J,Yang HM,Gui YT,Wang J,Cai ZM. Wholegenome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet,2013,45(12):1459–1463.
[11]张丽,陈杰. 全基因组关联研究及第二代测序技术在胰腺癌中的相关研究. 中华病理学杂志,2014,43(2):132–135.
[12]陈宾,马建婷,陈利玲,洪旭涛,唐晓婧,陈枢青. 腹膜间皮瘤组织体细胞突变的分析. 浙江大学学报 (医学版),2013,42(4): 426–430.
[13]Jiang YH,Yuen RK,Jin X,Wang MB,Chen N,Wu XL,Ju J,Mei JP,Shi YJ,He MZ,Wang GB,Liang JQ,Wang Z,Cao DD,Carter MT,Chrysler C,Drmic IE,Howe JL,Lau L,Marshall CR,Merico D,Nalpathamkalam T,Thiruvahindrapuram B,Thompson A,Uddin M,Walker S,Luo J,Anagnostou E,Zwaigenbaum L,Ring RH,Wang J,Lajonchere C,Wang J,Shih A,Szatmari P,Yang HM,Dawson G,Li YR,Scherer SW. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet,2013,93(2):249–263.
[14]Lu SJ,Zong CH,Fan W,Yang MY,Li JS,Chapman AR,Zhu P,Hu XS,Xu LY,Yan LY,Bai F,Qiao J,Tang FC,Li RQ,Xie XS. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science,2012,338(6114): 1627–1630.
[15]Patel RK,Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE,2012,7(2): e30619.
[16]Yang X,Liu D,Liu F,Wu J,Zou J,Xiao X,Zhao FQ,Zhu BL. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics,2013,14: 33.
[17]Dai MH,Thompson RC,Maher C,Contreras-Galindo R,Kaplan MH,Markovitz DM,Omenn G,Meng F.NGSQC: cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics,2010,11(Suppl. 4): S7.
[18]Li H,Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics,2010,26(5): 589–595.
[19]Langmead B,Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat methods,2012,9(4): 357–359.
[20]Li RQ,Li YR,Kristiansen K,Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics,2008,24(5): 713–714.
[21]Li H,Handsaker B,Wysoker A,Fennell T,Ruan J,Homer N,Marth G,Abecasis G,Durbin R,1000 Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics,2009,25(16): 2078–2079.
[22]Koboldt DC,Zhang QY,Larson DE,Shen D,McLellan MD,Lin L,Miller CA,Mardis ER,Ding L,Wilson RK.VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res,2012,22(3): 568–576.
[23]Li RQ,Li YR,Fang XD,Yang HM,Wang J,Kristiansen K,Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res,2009,19(6):1124–1132.
[24]Chiang DY,Getz G,Jaffe DB,O'Kelly MJT,Zhao XJ,Carter SL,Russ C,Nusbaum C,Meyerson M,Lander ES.High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods,2009,6(1):99–103.
[25]Abyzov A,Urban AE,Snyder M,Gerstein M. CNVnator:an approach to discover,genotype,and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res,2011,21(6): 974–984.
[26]New methods for detecting Salmonella. Anal Chem,2000,72(11): 387A.
[27]Ivakhno S,Royce T,Cox AJ,Evers DJ,Cheetham RK,Tavare S. CNAseg--a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics,2010,26(24):3051–3058.
[28]Fan X,Abbott TE,Larson D,Chen K. BreakDancer:Identification of Genomic Structural Variation from Paired-End Read Mapping. Curr Protoc Bioinformatics,2014,DOI:10.1002/0471250953.bi1506s45.
[29]Layer RM,Chiang C,Quinlan AR,Hall IM. LUMPY: A probabilistic framework for structural variant discovery.Genome Biol,2014,15(6): R84.
[30]Wang JM,Mullighan CG,Easton J,Roberts S,Heatley SL,Ma J,Rusch MC,Chen K,Harris CC,Ding L,Holmfeldt L,Payne-Turner D,Fan X,Wei L,Zhao D,Obenauer JC,Naeve C,Mardis ER,Wilson RK,Downing JR,Zhang JH.CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods,2011,8(8): 652–654.
[31]Sindi S,Helman E,Bashir A,Raphael BJ. A geometric approach for classification and comparison of structural variants. Bioinformatics,2009,25(12): i222–i230.
[32]Zeitouni B,Boeva V,Janoueix-Lerosey I,Loeillet S,Legoix-né P,Nicolas A,Delattre O,Barillot E. SVDetect:a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics,2010,26(15): 1895–1896.
[33]Michaelson JJ,Shi YJ,Gujral M,Zheng HC,Malhotra D,Jin X,Jian MH,Liu GM,Greer D,Bhandari A,Wu WT,Corominas R,Peoples A,Koren A,Gore A,Kang SL,Lin GN,Estabillo J,Gadomski T,Singh B,Zhang K,Akshoomoff N,Corsello C,McCarroll S,Iakoucheva LM,Li YR,Wang J,Sebat J. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation.Cell,2012,151(7): 1431–1442.
[34]Liu YZ,Li BS,Tan RJ,Zhu XL,Wang YD. A gradient-boosting approach for filtering de novo mutations in parent-offspring trios. Bioinformatics,2014,30(13):1830–1836.
[35]Li BS,Chen W,Zhan XW,Busonero F,Sanna S,Sidore C,Cucca F,Kang HM,Abecasis GR. A likelihood-based framework for variant calling and de novo mutation detection in families. PLoS Genet,2012,8(10): e1002944.
[36]Ramu A,Noordam MJ,Schwartz RS,Wuster A,Hurles ME,Cartwright RA,Conrad DF. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods,2013,10(10): 985–987.
[37]Wang K,Li MY,Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res,2010,38(16): e164.
[38]Sana ME,Iascone M,Marchetti D,Palatini J,Galasso M,Volinia S. GAMES identifies and annotates mutations in next-generation sequencing projects. Bioinformatics,2011,27(1): 9–13.
[39]Bi C,Wu Jy,Jiang T,Liu Q,Cai Ws,Yu P,Cai T,Zhao M,Jiang YH,Sun ZS. Mutations of ANK3 identified by exome sequencing are associated with Autism susceptibility. Hum Mutat,2012,33(12): 1635–1638.
[40]Kessler RC,Berglund P,Demler O,Jin R,Merikangas KR,Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry,2005,62(6): 593–602.
[41]Kessler RC,Amminger GP,Aguilar-Gaxiola S,Alonso J,Lee S,Ustün TB. Age of onset of mental disorders: a review of recent literature. Curr Opin Psychiatr,2007,20(4): 359–364.
[42]Collins PY,Patel V,Joestl SS,March D,Insel TR,Daar AS,Bordin IA,Costello EJ,Durkin M,Fairburn C,Glass RI,Hall W,Huang YQ,Hyman SE,Jamison K,Kaaya S,Kapur S,Kleinman A,Ogunniyi A,Otero-Ojeda A,Poo MM,Ravindranath V,Sahakian BJ,Saxena S,Singer PA,Stein DJ,Anderson W,Dhansay MA,Ewart W,Phillips A,Shurin S,Walport M. Grand challenges in global mental health. Nature,2011,475(7354): 27–30.
[43]Crow JF. The origins,patterns and implications of human spontaneous mutation. Nat Rev Genet,2000,1(1): 40–47.
[44]Eyre-Walker A,Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet,2007,8(8):610–618.
[45]Veeramah KR,O'Brien JE,Meisler MH,Cheng XY,Dib-Hajj SD,Waxman SG,Talwar D,Girirajan S,Eichler EE,Restifo LL,Erickson RP,Hammer MF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet,2012,90(3): 502–510.
[46]Schuurs-Hoeijmakers JHM,Geraghty MT,Kamsteeg EJ,Ben-Salem S,de Bot ST,Nijhof B,van de Vondervoort IIGM,van der Graaf M,Nobau AC,Otte-Höller I,Vermeer S,Smith AC,Humphreys P,Schwartzentruber J,FORGE Canada Consortium,Ali BR,Al-Yahyaee SA,Tariq S,Pramathan T,Bayoumi R,Kremer HPH,van de Warrenburgbp,van den Akker WM,Gilissen C,Veltman JA,Janssen IM,Vulto-van Silfhout AT,van der Velde-Visser S,Lefeber DJ,Diekstra A,Erasmus CE,Willemsen MA,Vissers LE,Lammens M,van Bokhoven H,Brunner HG,Wevers RA,Schenck A,Al-Gazali L,de Vries BB,de Brouwer AP. Mutations in DDHD2,Encoding an Intracellular Phospholipase A(1),Cause a Recessive Form of Complex Hereditary Spastic Paraplegia.Am J Hum Genet,2012,91(6): 1073–1081.
[47]Barcia G,Fleming MR,Deligniere A,Gazula VR,Brown MR,Langouet M,Chen HJ,Kronengold J,Abhyankar A,Cilio R,Nitschke P,Kaminska A,Boddaert N,Casanova JL,Desguerre I,Munnich A,Dulac O,Kaczmarek LK,Colleaux L,Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet,2012,44(11):1255–1259.
[48]Hamosh A,Scott AF,Amberger JS,Bocchini CA,McKusick VA. Online Mendelian Inheritance in Man(OMIM),a knowledgebase of human genes and genetic disorders. Nucleic Acids Res,2005,33(Database issue):D514–D517.
[49]Blake JA,Bult CJ,Eppig JT,Kadin JA,Richardson JE,Mouse Genome Database Group. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res,2014,42(Database issue): D810–D817.
[50]Forbes SA,Bindal N,Bamford S,Cole C,Kok CY,Beare D,Jia MM,Shepherd R,Leung K,Menzies A,Teague JW,Campbell PJ,Stratton MR,Futreal PA. COSMIC:mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res,2011,39(Suppl 1): D945–D950.
[51]Landrum MJ,Lee JM,Riley GR,Jang W,Rubinstein WS,Church DM,Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res,2014,42(Database issue):D980–D985.
[52]Stenson PD,Mort M,Ball EV,Shaw K,Phillips AD,Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics,diagnostic testing and personalized genomic medicine. Hum Genet,2014,133(1): 1–9.
[53]Khurana E,Fu Y,Colonna V,Mu XJ,Kang HM,Lappalainen T,Sboner A,Lochovsky L,Chen JM,Harmanci A,Das J,Abyzov A,Balasubramanian S,Beal K,Chakravarty D,Challis D,Chen Y,Clarke D,Clarke L,Cunningham F,Evani US,Flicek P,Fragoza R,Garrison E,Gibbs R,Gümüs ZH,Herrero J,Kitabayashi N,Kong Y,Lage K,Liluashvili V,Lipkin SM,MacArthur DG,Marth G,Muzny D,Pers TH,Ritchie GR,Rosenfeld JA,Sisu C,Wei XM,Wilson M,Xue YL,Yu FL,1000 Genomes Project Consortium,Dermitzakis ET,Yu HY,Rubin MA,Tyler-Smith C,Gerstein M. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science,2013,342(6154),DOI:10.1126/science.1235587.
[54]Li H. Aligning sequence reads,clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv:13033997,2013.
[55]O'Roak BJ,Deriziotis P,Lee C,Vives L,Schwartz JJ,Girirajan S,Karakoc E,Mackenzie AP,Ng SB,Baker C,Rieder MJ,Nickerson DA,Bernier R,Fisher SE,Shendure J,Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations.Nat Genet,2011,43(6): 585–589.
[56]Lee H,Lin MCA,Kornblum HI,Papazian DM,Nelson SF.Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet,2014,23(13): 3481–3489.
[57]Hamdan FF,Daoud H,Patry L,Dionne-Laporte A,Spiegelman D,Dobrzeniecka S,Rouleau GA,Michaud JL.Parent-child exome sequencing identifiesa de novo truncating mutation in TCF4 in non-syndromic intellectual disability. Clin Genet,2013,83(2): 198–200.
[58]Coordinators NR. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res,2014,42(Database issue): D7–D17.
[59]Fu W,O'Connor TD,Jun G,Kang HM,Abecasis G,Leal SM,Gabriel S,Rieder MJ,Altshuler D,Shendure J,Nickerson DA,Bamshad MJ,Project NES,Akey JM.Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature,2013,493(7431): 216–220.
[60]1000 Genomes Project Consortium,Abecasis GR,Auton A,Brooks LD,DePristo MA,Durbin RM,Handsaker RE,Kang HM,Marth GT,McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature,2012,491(7422): 56–65.
[61]Kumar P,Henikoff S,Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc,2009,4(7): 1073–1081.
[62]Adzhubei IA,Schmidt S,Peshkin L,Ramensky VE,Gerasimova A,Bork P,Kondrashov AS,Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods,2010,7(4): 248–249.
[63]Davydov EV,Goode DL,Sirota M,Cooper GM,Sidow A,Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++.PLoS Comput Biol,2010,6(12): e1001025.
[64]Chun S,Fay JC. Identification of deleterious mutations within three human genomes. Genome Res,2009,19(9):1553–1561.
[65]Maurano MT,Humbert R,Rynes E,Thurman RE,Haugen E,Wang H,Reynolds AP,Sandstrom R,Qu HZ,Brody J,Shafer A,Neri F,Lee K,Kutyavin T,Stehling-Sun S,Johnson AK,Canfield TK,Giste E,Diegel M,Bates D,Hansen RS,Neph S,Sabo PJ,Heimfeld S,Raubitschek A,Ziegler S,Cotsapas C,Sotoodehnia N,Glass I,Sunyaev SR,Kau R,Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science,2012,337(6099): 1190–1195.
[66]Khurana E,Fu Y,Colonna V,Mu XJ,Kang HM,Lappalainen T,Sboner A,Lochovsky L,Chen JM,Harmanci A,Das J,Abyzov A,Balasubramanian S,Beal K,Chakravarty D,Challis D,Chen Y,Clarke D,Clarke L,Cunningham F,Evani US,Flicek P,Fragoza R,Garrison E,Gibbs R,Gümüş ZH,Herrero J,Kitabayashi N,Kong Y,Lage K,Liluashvili V,Lipkin SM,MacArthur DG,Marth G,Muzny D,Pers TH,Ritchie GRS,Rosenfeld JA,Sisu C,Wei XM,Wilson M,Xue YL,Yu FL,1000 Genomes Project Consortium,Dermitzakis ET,Yu HY,Rubin MA,Tyler-Smith C,Gerstein M. Integrative annotation of variants from 1092 humans: Application to cancer genomics. Science,2013,342(6154),DOI:10.1126/science.1235587.
[67]Pleasance ED,Cheetham RK,Stephens PJ,McBride DJ,Humphray SJ,Greenman CD,Varela I,Lin ML,Ordóñez GR,Bignell GR,Ye K,Alipaz J,Bauer MJ,Beare D,Butler A,Carter RJ,Chen LN,Cox AJ,Edkins S,Kokko-Gonzales PI,Gormley NA,Grocock RJ,Haudenschild CD,Hims MM,James T,Jia MM,Kingsbury Z,Leroy C,Marshall J,Menzies A,Mudie LJ,Ning ZM,Royce T,Schulz-Trieglaff OB,Spiridou A,Stebbings LA,Szajkowski L,Teague J,Williamson D,Chin L,Ross MT,Campbell PJ,Bentley DR,Futreal PA,Stratton MR.A comprehensive catalogue of somatic mutations from a human cancer genome. Nature,2009,463(7278): 191–196.
[68]Pleasance ED,Stephens PJ,O’Meara S,McBride DJ,Meynert A,Jones D,Lin ML,Beare D,Lau KW,Greenman C,Varela I,Nik-Zainal S,Davies HR,Ordoñez GR,Mudie LJ,Latimer C,Edkins S,Stebbings L,Chen L,Jia M,Leroy C,Marshall J,Menzies A,Butler A,Teague JW,Mangion J,Sun YA,McLaughlin SF,Peckham HE,Tsung EF,Costa GL,Lee CC,Minna JD,Gazdar A,Birney E,Rhodes MD,McKernan KJ,Stratton MR,Futreal PA,Campbell PJ. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature,2010,463(7278): 184–190.
[69]Lee W,Jiang ZS,Liu JF,Haverty PM,Guan YH,Stinson J,Yue P,Zhang Y,Pant KP,Bhatt D,Ha C,Johnson S,Kennemer MI,Mohan S,Nazarenko I,Watanabe C,Sparks AB,Shames DS,Gentleman R,de Sauvage FJ,Stern H,Pandita A,Ballinger DG,Drmanac R,Modrusan Z,Seshagiri S,Zhang ZM. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature,2010,465(7297): 473–477.
[70]Lawrence MS,Stojanov P,Polak P,Kryukov GV,Cibulskis K,Sivachenko A,Carter SL,Stewart C,Mermel CH,Roberts SA,Kiezun A,Hammerman PS,McKenna A,Drier Y,Zou LH,Ramos AH,Pugh TJ,Stransky N,Helman E,Kim J,Sougnez C,Ambrogio L,Nickerson E,Shefler E,Cortés ML,Auclair D,Saksena G,Voet D,Noble M,DiCara D,Lin P,Lichtenstein L,Heiman DI,Fennell T,Imielinski M,Hernandez B,Hodis E,Baca S,Dulak AM,Lohr J,Landau DA,Wu CJ,Melendez-Zajgla J,Hidalgo-Miranda A,Koren A,McCarroll SA,Mora J,Lee RS,Crompton B,Onofrio R,Parkin M,Winckler W,Ardlie K,Gabriel SB,Roberts CW,Biegel JA,Stegmaier K,Bass AJ,Garraway LA,Meyerson M,Golub TR,Gordenin DA,Sunyaev S,Lander ES,Getz G. Mutational heterogeneity in cancer and the search for new cancerassociated genes. Nature,2013,499(7457): 214–218.
[71]Puente XS,Pinyol M,Quesada V,Conde L,Ordóñez GR,Villamor N,Escaramis G,Jares P,Beà S,González-Díaz M,Bassaganyas L,Baumann T,Juan M,López-Guerra M,Colomer D,Tubio JM,López C,Navarro A,Tornador C,Aymerich M,Rozman M,Hernández JM,Puente DA,Freije JMP,Velasco G,Gutiérrez-Fernández A,Costa D,Carrió A,Guijarro S,Enjuanes A,Hernández L,Yagüe J.,icolás P,Romeo-Casabona CM,Himmelbauer H,Castillo E,Dohm JC,de Sanjosé S,Piris MA,de Alava E,San Miguel J,Royo R,GelpíJL,Torrents D,Orozco M,Pisano DG,Valencia A,Guigó R,Bayés M,Heath S,Gut M,Klatt P,Marshall J,Raine K,Stebbings LA,Futreal PA,Stratton MR,Campbell PJ,Gut I,López-Guillermo A,Estivill X,Montserrat E,López-Otín C,Campo E.Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature,2011,475(7354):101–105.
[72]Roberts KG,Morin RD,Zhang JH,Hirst M,Zhao YJ,Su XP,Chen SC,Payne-Turner D,Churchman ML,Harvey RC,Chen X,Kasap C,Yan CH,Becksfort J,Finney RP,Teachey DT,Maude SL,Tse K,Moore R,Jones S,Mungall K,Birol I,Edmonson MN,Hu Y,Buetow KE,Chen IM,Carroll WL,Wei L,Ma J,Kleppe M,Levine RL,Garcia-Manero G,Larsen E,Shah NP,Devidas M,Reaman G,Smith M,Paugh SW,Evans WE,Grupp SA,Jeha S,Pui CH,Gerhard DS,Downing JR,Willman CL,Loh M,Hunger SP,Marra MA,Mullighan CG. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia.Cancer Cell,2012,22(2): 153–166.
[73]Wang K,Yuen ST,Xu J,Lee SP,Yan HH,Shi ST,Siu HC,Deng S,Chu KM,Law S,Chan KH,Chan AS,Tsui WY,Ho SL,Chan AK,Man JL,Foglizzo V,Ng MK,Chan AS,Ching YP,Cheng GH,Xie T,Fernandez J,Li VS,Clevers H,Rejto PA,Mao M,Leung SY. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet,2014,46(6): 573–582.
[74]Holmfeldt L,Wei L,Diaz-Flores E,Walsh M,Zhang JH,Ding L,Payne-Turner D,Churchman M,Andersson A,Chen SC,McCastlain K,Becksfort J,Ma J,Wu G,Patel SN,Heatley SL,Phillips LA,Song G,Easton J,Parker M,Chen X,Rusch M,Boggs K,Vadodaria B,Hedlund E,Drenberg C,Baker S,Pei D,Cheng C,Huether R,Lu C,Fulton RS,Fulton LL,Tabib Y,Dooling DJ,Ochoa K,Minden M,Lewis ID,To LB,Marlton P,Roberts AW,Raca G,Stock W,Neale G,Drexler HG,Dickins RA,Ellison DW,Shurtleff SA,Pui CH,Ribeiro RC,Devidas M,Carroll AJ,Heerema NA,Wood B,Borowitz MJ,Gastier-Foster JM,Raimondi SC,Mardis ER,Wilson RK,Downing JR,Hunger SP,Loh ML,Mullighan CG. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet,2013,45(3): 242–252.
[75]Turajlic S,Furney SJ,Lambros MB,Mitsopoulos C,Kozarewa I,Geyer FC,MacKay A,Hakas J,Zvelebil M,Lord CJ,Ashworth A,Thomas M,Stamp G,Larkin J,Reis-Filho JS,Marais R. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res,2012,22(2): 196–207.
[76]Brastianos PK,Horowitz PM,Santagata S,Jones RT,McKenna A,Getz G,Ligon KL,Palescandolo E,Van Hummelen P,Ducar MD,Raza A,Sunkavalli A,Macconaill LE,Stemmer-Rachamimov AO,Louis DN,Hahn WC,Dunn IF,Beroukhim R. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet,2013,45(3): 285–289.
[77]Nik-Zainal S,Alexandrov LB,Wedge DC,Van Loo P,Greenman CD,Raine K,Jones D,Hinton J,Marshall J,Stebbings LA,Menzies A,Martin S,Leung K,Chen L,Leroy C,Ramakrishna M,Rance R,Lau KW,Mudie LJ,Varela I,McBride DJ,Bignell GR,Cooke SL,Shlien A,Gamble J,Whitmore I,Maddison M,Tarpey PS,Davies HR,Papaemmanuil E,Stephens PJ,McLaren S,Butler AP,Teague JW,Jönsson G,Garber JE,Silver D,Miron P,Fatima A,Boyault S,Langerød A,Tutt A,Martens JW,Aparicio SA,Borg Å,Salomon AV,Thomas G,Børresen-Dale AL,Richardson AL,Neuberger MS,Futreal PA,Campbell PJ,Stratton MR; Breast Cancer Working Group of the International Cancer Genome Consortium. Mutational processes molding the genomes of 21 breast cancers. Cell,2012,149(5): 979–993.
[78]Robinson G,Parker M,Kranenburg TA,Lu C,Chen X,Ding L,Phoenix TN,Hedlund E,Wei L,Zhu XY,Chalhoub N,Baker SJ,Huether R,Kriwacki R,Curley N,Thiruvenkatam R,Wang J,Wu G,Rusch M,Hong X,Becksfort J,Gupta P,Ma J,Easton J,Vadodaria B,Onar-Thomas A,Lin T,Li S,Pounds S,Paugh S,Zhao D,Kawauchi D,Roussel MF,Finkelstein D,Ellison DW,Lau CC,Bouffet E,Hassall T,Gururangan S,Cohn R,Fulton RS,Fulton LL,Dooling DJ,Ochoa K,Gajjar A,Mardis ER,Wilson RK,Downing JR,Zhang J,Gilbertson RJ.Novel mutations target distinct subgroups of medulloblastoma. Nature,2012,488(7409): 43–48.
[79]Sato Y,Yoshizato T,Shiraishi Y,Maekawa S,Okuno Y,Kamura T,Shimamura T,Sato-Otsubo A,Nagae G,Suzuki H,Nagata Y,Yoshida K,Kon A,Suzuki Y,Chiba K,Tanaka H,Niida A,Fujimoto A,Tsunoda T,Morikawa T,Maeda D,Kume H,Sugano S,Fukayama M,Aburatani H,Sanada M,Miyano S,Homma Y,Ogawa S. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet,2013,45(8): 860–867.
[80]Han JY,Lee YS,Kim BC,Lee GK,Lee S,Kim EH,Kim HM,Bhak J. Whole-genome analysis of a patient with early-stage small-cell lung cancer. Pharmacogenomics J,2014,DOI:10.1038/tpj.2014.17.
[81]Mohan S,Heitzer E,Ulz P,Lafer I,Lax S,Auer M,Pichler M,Gerger A,Eisner F,Hoefler G. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet,2014,10(3): e1004271.
[82]Demeure MJ,Aziz M,Rosenberg R,Gurley SD,Bussey KJ,Carpten JD. Whole-genome Sequencing of an Aggressive BRAF Wild-type Papillary Thyroid Cancer Identified EML4–ALK Translocation as a Therapeutic Target. World J Surg,2014,38(6): 1296–1305.
[83]Talkowski ME,Rosenfeld JA,Blumenthal I,Pillalamarri V,Chiang C,Heilbut A,Ernst C,Hanscom C,Rossin E,Lindgren AM,Pereira S,Ruderfer D,Kirby A,Ripke S,Harris DJ,Lee JH,Ha K,Kim HG,Solomon BD,Gropman AL,Lucente D,Sims K,Ohsumi TK,Borowsky ML,Loranger S,Quade B,Lage K,Miles J,Wu BL,Shen Y,Neale B,Shaffer LG,Daly MJ,Morton CC,Gusella JF.Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell,2012,149(3): 525–537.
[84]Kong A,Frigge ML,Masson G,Besenbacher S,Sulem P,Magnusson G,Gudjonsson SA,Sigurdsson A,Jonasdottir A,Jonasdottir A,Wong WS,Sigurdsson G,Walters GB,Steinberg S,Helgason H,Thorleifsson G,Gudbjartsson DF,Helgason A,Magnusson OT,Thorsteinsdottir U,Stefansson K. Rate of de novo mutations and the importance of father's age to disease risk. Nature,2012,488(7412): 471–475.
[85]Chiu RW. Noninvasive prenatal testing by maternal plasma DNA analysis: Current practice and future applications. Scand J Clin Lab Invest Suppl,2014,244:48–53.
[86]Chiu RWK,Chan KCA,Gao Y,Lau VYM,Zheng WL,Leung TY,Foo CHF,Xie B,Tsui NBY,Lun FMF,Zee BCY,Lau TK,Cantor CR,Lo YMD. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma.Proc Natl Acad Sci U S A,2008,105(51): 20458–20463.
[87]Lau TK,Jiang FM,Stevenson RJ,Lo TK,Chan LW,Chan MK,Lo PSS,Wang W,Zhang HY,Chen F,Choy KW.Secondary findings from non-invasive prenatal testing for common fetal aneuploidies by whole genome sequencing as a clinical service. Prenat Diagn,2013,33(6):602–608.
[88]Leary RJ,Sausen M,Kinde I,Papadopoulos N,Carpten JD,Craig D,O'Shaughnessy J,Kinzler KW,Parmigiani G,Vogelstein B,Diaz LA Jr,Velculescu VE. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med,2012,4(162): 162ra154.
[89]Choi M,Scholl UI,Ji WZ,Liu TW,Tikhonova IR,Zumbo P,Nayir A,Bakkaloğlu A,Özen S,Sanjad S,Nelson-Williams C,Farhi A,Mane S,Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA,2009,106(45):19096–19101.
[90]Bonnefond A,Durand E,Sand O,De Graeve F,Gallina S,Busiah K,Lobbens S,Simon A,Bellanné-Chantelot C,Létourneau L,Scharfmann R,Delplanque J,Sladek R,Polak M,Vaxillaire M,Froguel P. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS ONE,2010,5(10): e13630.
[91]Worthey EA,Mayer AN,Syverson GD,Helbling D,Bonacci BB,Decker B,Serpe JM,Dasu T,Tschannen MR,Veith RL,Basehore MJ,Broeckel U,Tomita-Mitchell A,Arca MJ,Casper JT,Margolis DA,Bick DP,Hessner MJ,Routes JM,Verbsky JW,Jacob HJ,Dimmock DP. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med,2011,13(3):255–262.
[92]Montenegro G,Powell E,Huang J,Speziani F,Edwards YJK,Beecham G,Hulme W,Siskind C,Vance J,Shy M,Züchner S. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol,2011,69(3): 464–470.
[93]Lo YM. Fetal DNA in maternal plasma: biology and diagnostic applications. Clin Chem,2000,46(12): 1903–1906.
[94]Puckelwartz MJ,Pesce LL,Nelakuditi V,Dellefave-Castillo L,Golbus JR,Day SM,Cappola TP,Dorn II GW,Foster IT,McNally EM. Supercomputing for the parallelization of whole genome analysis. Bioinformatics,2014,30(11): 1508–1513.
[95]Tischler G,Leonard S. Biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol Med,2014,9: 13.
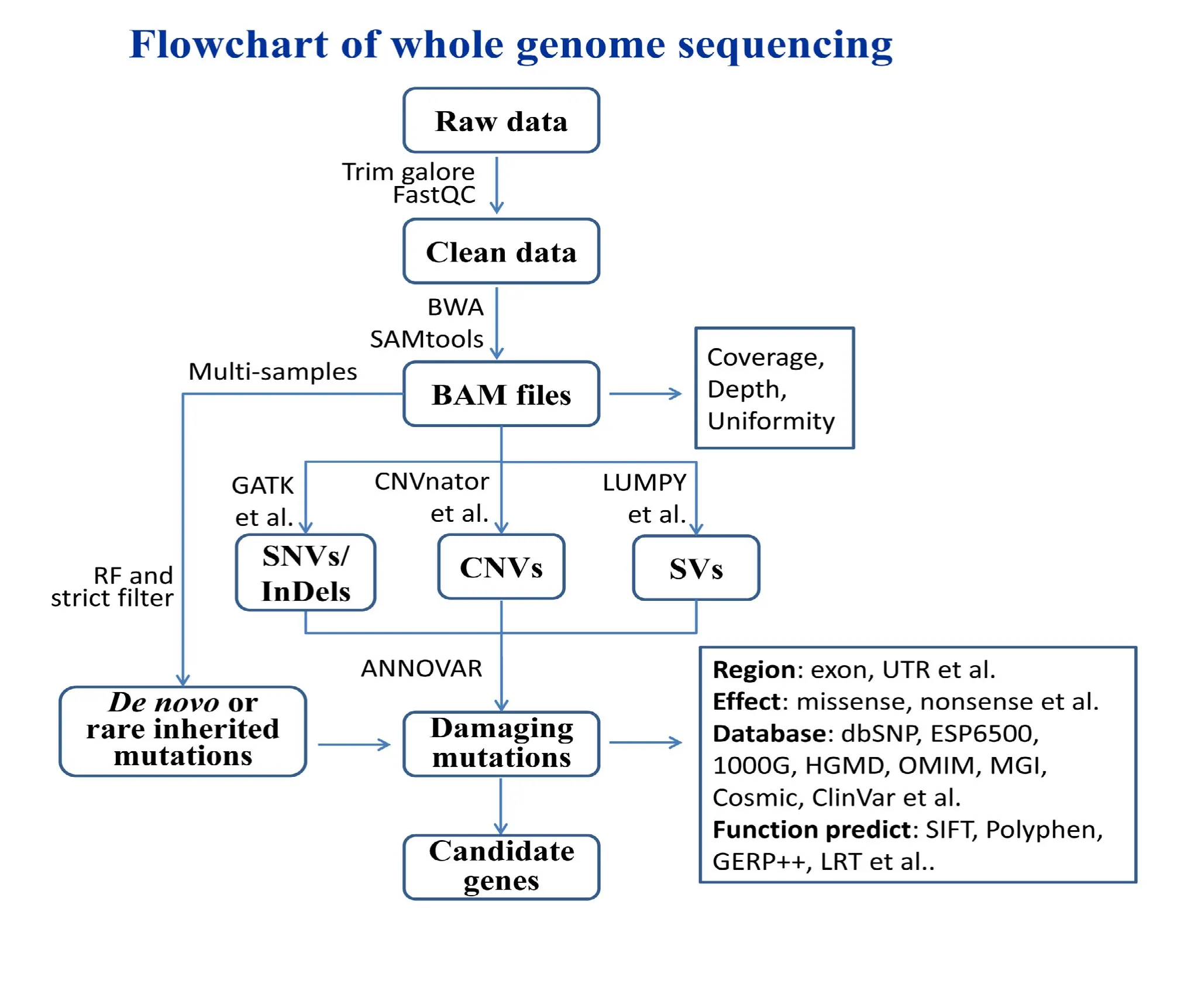
附图1
猜你喜欢
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
中成药(2018年7期)2018-08-04
