
一个中国遗传性血色病家系致病基因的突变分析
2014-05-10 01:26:22李元丰张红星张海涛彭晓波白丽丽贺福初邱泽武周钢桥
遗传 2014年11期
李元丰,张红星,张海涛,彭晓波,白丽丽,贺福初,邱泽武,周钢桥
1.中国人民解放军军事医学科学院放射与辐射医学研究所,北京蛋白质组研究中心,蛋白质组学国家重点实验室,北京 102206;
2.中国人民解放军第三〇七医院中毒救治,北京 100071;
3.蛋白质药物国家工程研究中心,北京 102206;
4.国家蛋白质科学中心(北京),北京 102206
遗传性血色病(Hereditary hemochromatosis, HHC)是一种罕见的常染色体隐性遗传病,最常见于北欧白种人群,发病率可达 0.5%[1]。该病在中国人群中的发病率较低,但至今尚无确切的流行病学统计数据。HHC患者由于遗传缺陷,导致过多的铁进入血循环,进而引起铁逐渐在肝脏、胰腺 和心脏等多种器官中沉积,并最终损伤器官功能。HHC的临床表现为皮肤色素沉着、肝硬化、糖尿病、心律失常、心功能衰竭、垂体损伤、睾丸萎缩和关节疾病等。
目前已发现有5个基因的突变可能导致遗传性血色病,分别是 HAMP、HJV(HFE2)、TFR2、FPN和HFE[2]。根据不同的遗传病因,可将HHC分为4种类型[3]:1型为HFE突变所致的血色病,故又被称为HFE相关血色病,绝大多数遗传性血色病属于该类型;2型为HJV基因和HAMP基因突变所致的血色病,其中前者被进一步划分为2A型,后者被划分为2B型;3型为TFR2基因突变所致的血色病;4型为FPN基因突变所致的血色病。
本研究收集了我国安徽省的一个近亲婚配家系,并使用全外显子组测序技术检测了先证者及其 1个无症状堂(表)弟的外周血基因组DNA上的罕见突变,对该家系的致病基因进行了定位。
1 对象和方法
1.1 血色病患者及所在家系的临床和社会人口学资料
患者(先证者)来自安徽省涡阳县,男性,汉族,29岁。2012年12月因“出现皮肤发黑、腹胀、胸闷、憋气等症状”就诊。实验室检查发现:铁蛋白含量为1650 ng/mL、总胆红素为46.6 µmol/L、直接胆红素为20.6 µmol/L。临床诊断为心肌病,全心功能不全IV级,房室传导阻塞,疑为血色病。进一步诊治及既往史调查发现,该病人性功能丧失已10年,脾脏肿大,患2型糖尿病,不排除肝硬化的可能性。上述临床表现及检查结果提示该病人患有血色病。
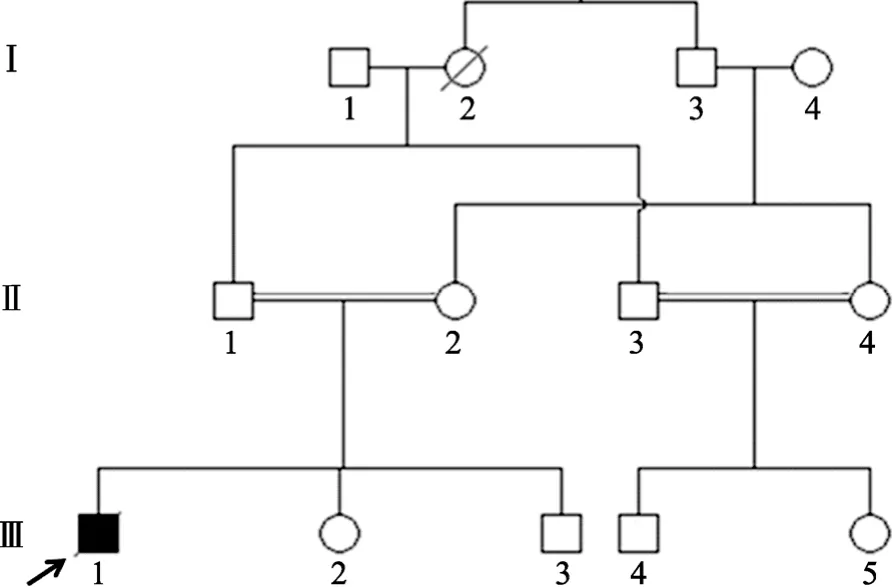
图1 遗传性血色病家系示意图
患者父母为姑表近亲结婚(图1),该家系的遗传关系均得到确认。家系中其他成员暂未出现明显的血色病症状。本研究经军事医学科学院放射与辐射医学研究所医学伦理委员会批准执行,所有受试者均签署了知情同意书。
1.2 方法
1.2.1 外周血基因组DNA的提取
抽取血色病患者(Ⅲ-1)和其他4名家系成员(Ⅲ-2、Ⅲ-3、Ⅲ-4和Ⅲ-5)的外周静脉全血,用常规得饱和酚、氯仿法抽提基因组 DNA,乙醇沉淀保存待用。该家系的其余成员未提供外周血样。
1.2.2 HFE c.845G>A突变的检测与分析
为检测先证者是否携带与血色病最为密切相关的 HFE基因的常见致病 c.845G>A(p.C282Y,该突变导致HFE蛋白第282位半胱氨酸 C被酪氨酸Y取代[p.C282Y]),本文采用 PCR扩增、产物纯化和Sanger测序法对先证者的基因组 DNA进行了突变分析。PCR扩增覆盖突变所在的基因组区域,上游引物为:5′-GGGTATTTCCTTCCTCCAACC-3′,下游引物为:5′-AATTACCTCCTCAGGCACTCC-3′。测序结果与野生型 HFE基因的标准序列(GenBank:NC_000006.12)比对,以确定该病例是否携带HFE c.845G>A突变。
1.2.3 全外显子组测序
本文对先证者(Ⅲ-1)和家系成员Ⅲ-4的基因组DNA进行了外显子区域的捕获和高通量测序。外显子区域捕获使用的试剂盒为 SureSelect人全外显子V5 + UTR(Agilent公司),大约覆盖人类基因组70 Mb区域。使用高通量测序仪Hiseq 2000进行双端100 bp的高通量测序(Illumina公司)。
1.2.4 序列比对、变异检测和注释
使用 Trimmomatic软件(v0.32)去除低质量的高通量测序数据,使用 BWA软件(v0.5.9)将高质量的测序数据与参考基因组(hg19)进行比对,使用GATK软件(v2.8-1)检测单碱基变异(SNV)和插入/缺失(Indel)。使用ANNOVAR对变异进行注释。保留目标区域中能改变氨基酸的突变(包括错义突变、无义突变、基因编码区内微小的插入缺失和可变剪接区的突变),并去除数据库中已知的单核苷酸多态性(Single-nucleotide polymorphism, SNP),包括dbSNP135数据库中的常见 SNP、千人基因组计划中亚洲人的SNP以及NHLBI外显子组测序项目中的常见SNP。通过以上分析、过滤和质量控制,最终获得个体中携带的具有潜在功能意义的罕见突变。
1.2.5 突变危害性的预测
使用SIFT(http://sift.jcvi.org/www/SIFT_chr_coords_submit.html)和PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/bgi.shtml)软件,预测突变对蛋白质功能的影响程度。仅当两种软件均预测同一突变对蛋白质的功能影响较大时,才认定该突变具有较强的危害性。
2 结果与分析
2.1 病例中未检测到HFE基因c.845G>A突变
前人已有报道,HFE基因上的纯合突变或复合杂合型突变是导致遗传性血色病的常见原因,在遗传性血色病患者中,HFE基因 c.845G>A纯合突变发生频率最高(该突变可导致HFE蛋白发生p.C282Y的改变),约占遗传性血色病患者总数的80%~85%[4]。通过 Sanger测序,发现血色病患者(Ⅲ-1)并不携带p.C282Y突变,从而排除该患者所患疾病是由p.C282Y纯合突变引起的,从而提示需要通过进一步的检测,以确定该血色病患者的其它致病突变。
2.2 全外显子组测序的深度及覆盖度
本文对先证者(Ⅲ-1)及另一家系成员(Ⅲ-4)的外周血基因组DNA进行了全外显子组测序。与参考基因组(hg19)比对后的结果显示,血色病患者Ⅲ-1及家系成员Ⅲ-4的平均测序深度分别达到 101.2×和111.7×,目标区域在 10×以上的比例达到 96%以上,说明全基因组范围内的编码区和非翻译区(Untranslated region,UTR)基本被有效覆盖(表1)。
2.3 在先证者中检测到 HJV(HFE2)基因存在两个纯合突变
通过生物信息学分析发现,该血色病患者在多个基因上携带纯合或杂合突变。在目前已知的与HHC相关的5个基因(HAMP、HJV、TFR2、FPN和HFE)中,HAMP、TFR2、FPN和 HFE未发生基因突变。但是,在 HJV基因上发生了两个纯合突变,分别为c.G18C和c.GC962_963AA,通过Sanger测序进一步证实了这两个纯合突变(图2A和2B)。其中,第一个突变能够引起HJV蛋白第6位谷氨酰胺被组氨酸取代(p.Q6H)。但是,通过生物信息学预测,发现该位点保守性差,突变引起的危害性小(SIFT和PolyPhen-2软件预测均提示危害性较小),因此推测该突变不是致病性突变。第二个突变为连续的两个碱基突变,引起HJV蛋白的翻译在第321位被提前终止(p.C321X),该突变造成HJV蛋白缺少尾部的糖基化磷脂酰肌醇(Glycosilphosphatidylinositol,GPI)锚定结构域,具有很强的危害性。此外,通过检查本实验室内部的 148个不患血色病的个体的高通量测序数据集,未能发现c.G18C和c.GC962_963AA这两个突变。由此推测,HJV基因上的c.GC962_963AA(p.C321X)纯合突变可能是血色病患者Ⅲ-1的致病性突变。

表1 测序深度统计
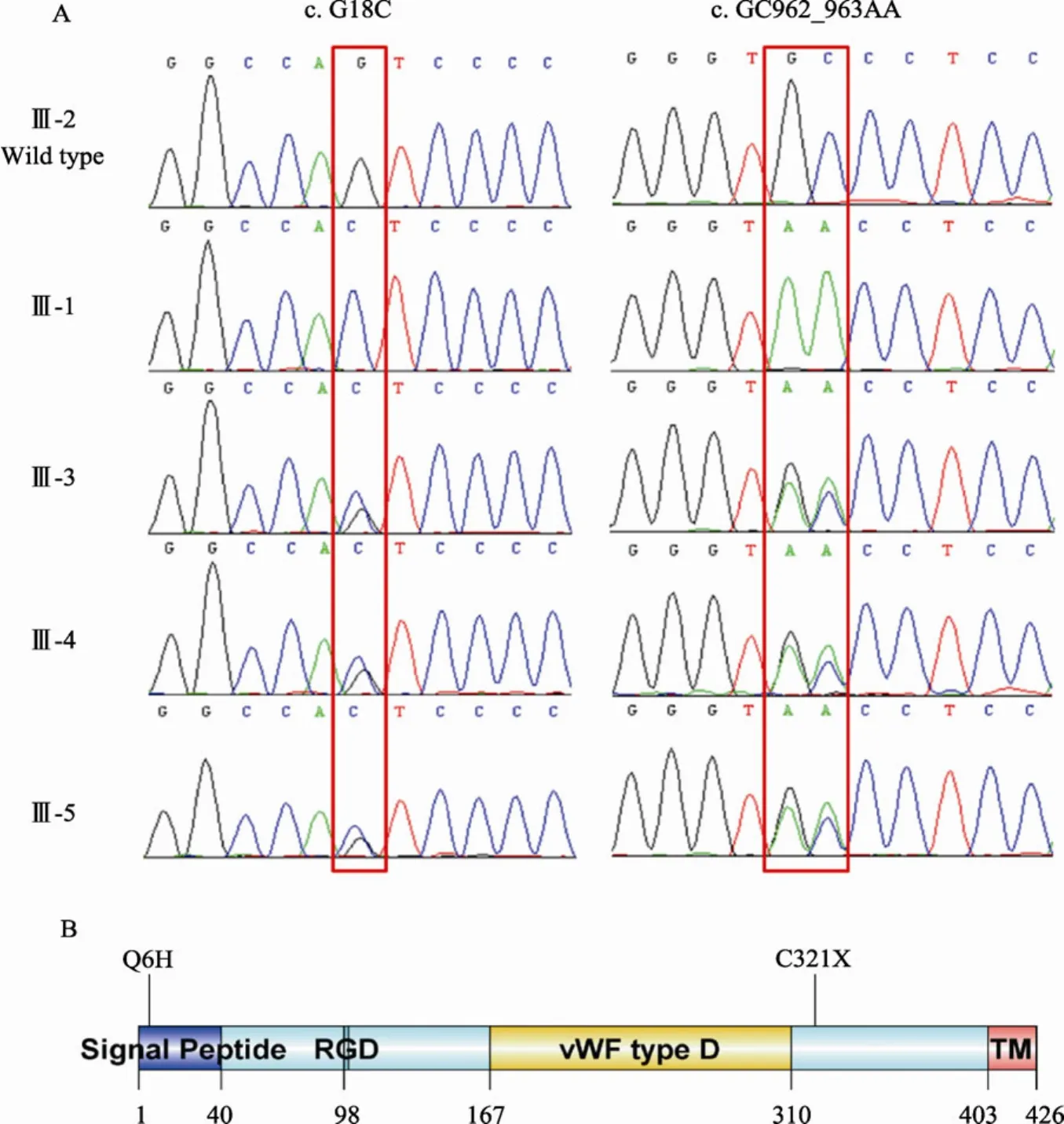
图2 HJV基因的突变
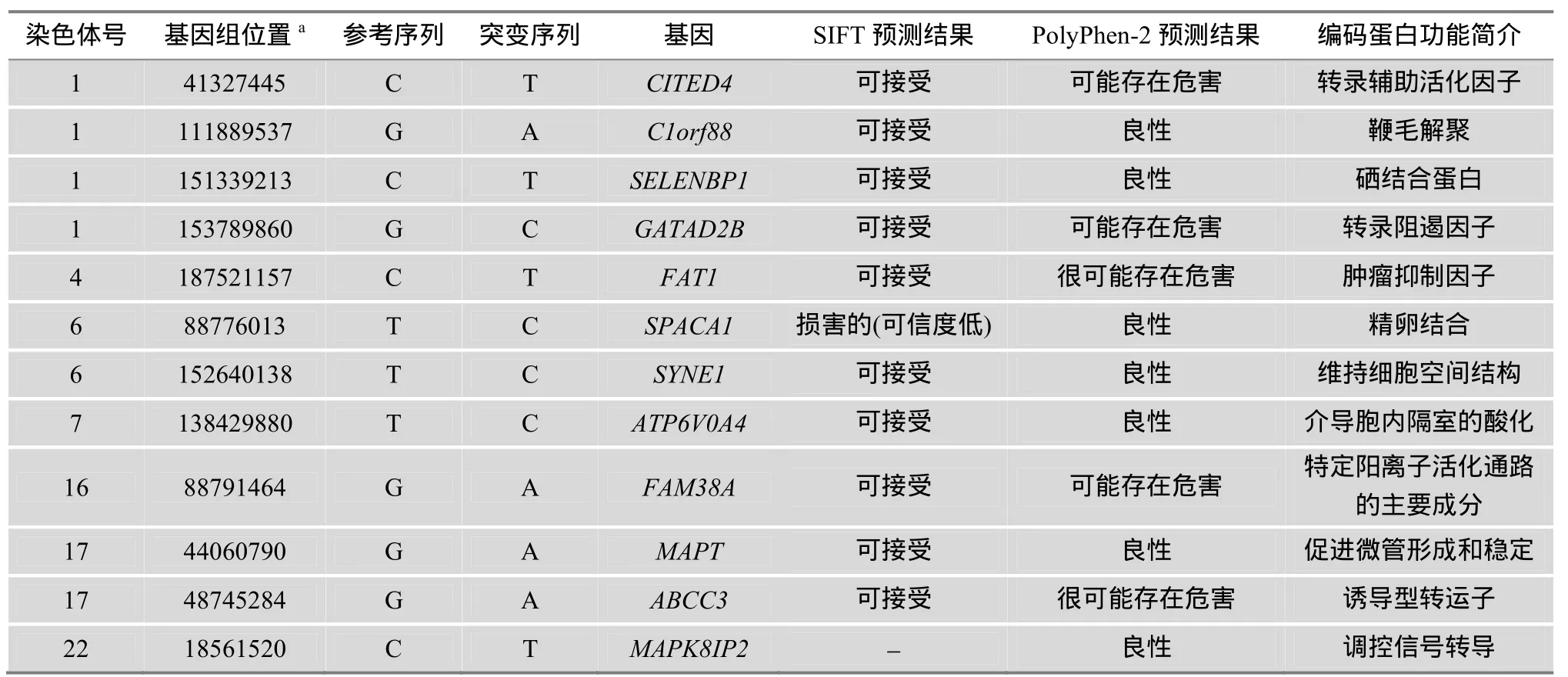
表2 病例中携带的纯合突变(除HJV基因突变外)
2.4 先证者的其它纯合突变
除目前已知的与遗传性血色病相关的 5个基因外,其他基因上的突变是否可能是血色病患者Ⅲ-1的致病突变?为此,本文进一步分析了该血色病患者携带的所有突变。考虑到该患者的父母为近亲结婚,推断该患者更有可能是由相关基因上的纯合突变引起的,而非复合杂合型突变,故以下仅关注纯合突变。
本文发现,除HJV基因上的2个纯合突变外,该患者携带其他 12个纯合突变(表 2)。通过生物信息学预测发现,这 12个突变的危害性均不强(两种预测软件同时预测突变危害性强时才认定该突变危害性强),且突变所在基因的功能与铁代谢之间的关系未见报道。因此,该患者携带的其他突变引发血色病的可能性比较低。
综上,HJV基因上的纯合突变c.GC962_963AA(p.C321X)很可能是血色病患者Ⅲ-1的致病性突变。由此,从遗传学上推测该患者所患疾病为2A型血色病。事实上,该患者的临床表现为发病年龄低,性功能丧失,全心功能不全,均符合2A型血色病的特点。
2.5 家系成员Ⅲ-4的全外显子组测序分析
家系成员Ⅲ-4(23岁)的转铁蛋白饱和度正常,但是该个体具有精子不液化等异常症状。为了尝试从遗传学上确定家系成员Ⅲ-4是否患有遗传性血色病,我们对该个体也进行了全外显子组测序。Ⅲ-4与血色病患者Ⅲ-1的亲缘关系非常近(图1),如果该个体也患有遗传性血色病,则一般情况下,该个体应携带与血色病患者Ⅲ-1一致的纯合突变 c.G18C和c.GC962_963AA(p.Q6H/p.C321X)。经分析,发现该个体确实携带与血色病患者Ⅲ-1一致的突变c.G18C和c.GC962_963AA,但均为杂合性突变(图2)。此外,该个体在HJV基因上不存在其它突变。该个体在其他4个已知的与遗传性血色病相关的基因上也不存在突变。因此,根据现有的遗传学知识,本文无法确定家系成员Ⅲ-4是否患有遗传性血色病。
2.6 其他家系成员的突变分析
本文进一步对该家系第Ⅲ代其余3名成员也进行了 c.G18C(p.Q6H)和 c.GC962_963AA(p.C321X)突变的检测。结果发现,家系成员Ⅲ-2为野生型纯合子,Ⅲ-3和Ⅲ-5为杂合子(图2)。目前,家系成员Ⅲ-3和Ⅲ-5表现型均正常,但考虑到二者年龄较低(24岁和 22岁),是否患有遗传性血色病尚需要进一步的观察。此外,本文注意到该家系第Ⅲ代的每一名成员均同时携带p.Q6H和p.C321X突变,或者既不携带p.Q6H也不携带p.C321X突变,提示p.Q6H与致病性的p.C321X突变呈连锁不平衡状态,处于同一等位基因上。
3 讨 论
遗传性血色病是一种罕见的常染色体隐性遗传病。目前已发现5个铁代谢相关基因(HAMP、HJV、TFR2、FPN和 HFE)上的突变能够导致血色病。其中,HFE基因上的突变最早被报道[6],且该基因上的突变是遗传性血色病发病的主要原因[7]。遗传性血色病患者在HFE蛋白上的突变主要为p.C282Y纯合突变、p.C282Y/p.H63D复合杂合型突变和p.C282Y/p.S65C复合杂合型突变[2]。这些突变在高加索人群中比较常见,但在我国人群中非常罕见[8],本研究中的血色病患者也不存在p.C282Y等突变。
本研究对同一家系的两名个体(其中一例为血色病患者)进行了全外显子组深度测序。结果显示,该家系中血色病患者的 HJV基因携带纯合突变p.C321X,提示该患者所患疾病为2A型血色病,其临床表型证实了该推测。
HJV mRNA主要在肝脏、心脏和骨骼肌中表达。其中,肝实质细胞内HJV的表达对铁调素(Hepcindin)的分泌具有重要的调节功能,进而维持机体内铁代谢的平衡[9]。HJV存在附着型(Membrane form,m-HJV)和可溶性(Soluble form,s-HJV)两种形式,当附着型 HJV尾部的糖基磷脂肌醇(GPI)锚定结构域被剪切掉后,可形成可溶性 HJV。两者对铁调素的调节功能是反向的。其中附着型HJV作为骨形成蛋白(Bone morphogenetic protein,BMP)的共受体,可通过BMP/SMAD通路上调铁调素的表达;而可溶性HJV可与附着型HJV竞争性结合BMP,导致铁调素的表达下调。近年来,在血色病患者体内已发现多种不同的HJV突变[10]。其中多种HJV突变可显著降低附着型HJV的表达水平。同时,研究还发现,携带HJV突变时,铁调素的表达异常降低[11],这进一步提示了HJV突变在血色病发生中的作用。
Huang等[12]曾报道过HJV突变p.C321X存在于一名中国年轻女性遗传性血色病患者中。不同于本研究的是,Huang等[12]在患者中检测到的突变p.C321X为杂合型,来源于其表型正常的母亲。该突变型等位基因与来源于表型正常的父亲的另一HJV突变型等位基因(p.I281T)形成复合杂合子,最终导致遗传性血色病。其中,p.I281T纯合突变已被报道能导致血色病[10]。p.C321X突变与以往报道的p.R326X突变的功能类似,二者均可引起HJV蛋白缺失尾部的糖基磷脂肌醇(GPI)锚定结构域,进而形成可溶性HJV,最终引发铁代谢异常[13]。
最近,Li等[14]报道了一名中国中年男性血色病患者携带杂合型HJV突变型等位基因p.C321X,该突变型等位基因来源于其健康的母亲。但是,该患者不携带其他的HJV基因突变,也不携带其他血色病相关基因(HAMP、TFR2、FPN和 HFE)的突变,这一结论与以往血色病是隐性遗传病的认识相悖。Li等进一步分析患者所在家系的其他成员,发现患者的一名姐姐和一名哥哥也从其母亲处继承了携带p.C321X突变的等位基因。有意思的是,患者的这名哥哥疑似患有血色病,而其姐姐身体健康。因为以往从未报道过健康男性携带p.C321X这一突变型等位基因,由此Li等认为基因和环境因素共同影响疾病的发生,携带一个异常HJV等位基因(p.C321X)的男性也有很大可能患血色病,但发病年龄可能比携带HJV基因纯合突变或复合杂合性突变的患者要晚;而携带一个异常等位基因p.C321X的女性则基本不会发病。
本研究中,血色病患者Ⅲ-1携带HJV纯合突变型等位基因 p.C321X,同时排除了其他基因上的突变可能导致该病。由此,本文首次报道了纯合 HJV突变型等位基因p.C321X可引起血色病。进一步对该家系其他成员进行突变检测后发现,该家系第Ⅲ代成员中的另外两名男性个体Ⅲ-3(24岁)和Ⅲ-4(23岁)均携带p.C321X杂合突变。这两名男性个体转铁蛋白饱和度正常,但是Ⅲ-4具有精子不液化等异常表型,且Ⅲ-3和Ⅲ-4年龄均较小。鉴于Li等[14]曾发现两名中国中年男性携带p.C321X杂合突变可导致血色病,因此不排除本研究中的两名青年男性今后罹患血色病的可能性。同时本文发现,该家系第Ⅲ代成员的一名女性个体Ⅲ-5也携带p.C321X杂合突变。结合 Huang等[12]和 Li等[14]的研究,推测该女性个体血色病的发病风险较低,但不排除其男性后代罹患血色病的可能性,需要密切关注。该家系第Ⅲ代成员的另一名女性个体Ⅲ-2是野生型纯合子,故该个体罹患遗传性血色病的风险较低。此外,由于血色病患者Ⅲ-1及其妹妹(Ⅲ-2)和弟弟(Ⅲ-3)分别为p.C321X突变型纯合子、野生型纯合子和杂合子,因此,虽然其父母没有提供外周血DNA用于本项研究,但是我们仍然能够根据孟德尔遗传定律推断其父母(Ⅱ-1和Ⅱ-2,表型正常)均携带一个 p.C321X突变的等位基因。这一点若能得到实验证实,将首次提示存在一例健康老年男性携带p.C321X杂合突变,从而提示只携带一个p.C321X突变等位基因不一定使男性患血色病。
综上,本研究首次在来自一个中国近亲结婚家系的血色病患者中检测到HJV p.C321X纯合突变。结合 Huang等[12]和 Li等[14]的研究,目前共在 3个中国血色病家系中发现了p.C321X突变,提示p.C321X这两个突变在中国人群中存在,但其发生频率较低。进一步在中国人群中特别是安徽和江苏一带的中国人群中筛查p.C321X突变,将有可能对中国血色病的致病机制和基因诊断研究发挥重要作用。
[1]Adams P, Brissot P, Powell LW.EASL international consensus conference on haemochromatosis.J Hepatol, 2000,33(3): 485–504.
[2]Pietrangelo A.Hereditary hemochromatosis: pathogenesis,diagnosis, and treatment.Gastroenterology, 2010, 139(2):393–408.e2.
[3]Franchini M.Hereditary iron overload: update on pathophysiology, diagnosis, and treatment.Am J Hematol,2006, 81(3): 202–209.
[4]European Association For The Study Of The Liver.EASL clinical practice guidelines for HFE hemochromatosis.J Hepatol, 2010, 53(1): 3–22.
[5]Ren J, Wen LP, Gao XJ, Jin CJ, Xue Y, Yao XB.DOG 1.0:illustrator of protein domain structures.Cell Res, 2009,19(2): 271–273.Simon M, Bourel M, Genetet B, Fauchet R.Idiopathic hemochromatosis.Demonstration of recessive transmission and early detection by family HLA typing.N Engl J Med, 1977, 297(19): 1017–1021.
[6]Distante S, Robson KJH, Graham-Campbell J, Arnaiz-Villena A, Brissot P, Worwood M.The origin and spread of the HFE-C282Y haemochromatosis mutation.Hum Genet, 2004, 115(4): 269–279.
[7]Lin A, Yan WH, Xu HH, Zhu M, Zhou MY.Analysis of the HFE gene (C282Y, H63D and S65C) mutations in a general Chinese Han population.Tissue Antigens, 2007, 70(3):252–255.
[8]Wang RH, Li CL, Xu XL, Zheng Y, Xiao CY, Zerfas P,Cooperman S, Eckhaus M, Rouault T, Mishra L, Deng CX.A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression.Cell Metab, 2005, 2(6):399–409.
[9]Camaschella C, Silvestri L.New and old players in the hepcidin pathway.Haematologica, 2008, 93(10): 1441–1444.
[10]Papanikolaou G, Samuels ME, Ludwig EH, MacDonald MLE, Franchini PL, Dubé MP, Andres L, MacFarlane J,Sakellaropoulos N, Politou M, Nemeth E, Thompson J,Risler J K, Zaborowska C, Babakaiff R, Radomski CC,Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G,Ganz T, Hayden MR, Goldberg YP.Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis.Nat Genet, 2004, 36(1): 77–82.
[11]Huang FW, Rubio-Aliaga I, Kushner JP, Andrews NC,Fleming MD.Identification of a novel mutation (C321X)in HJV.Blood, 2004, 104(7): 2176–2177.
[12]Silvestri L, Pagani A, Fazi C, Gerardi G, Levi S, Arosio P,Camaschella C.Defective targeting of hemojuvelin to plasma membrane is a common pathogenetic mechanism in juvenile hemochromatosis.Blood, 2007, 109(10):4503–4510.
[13]Li SF, Xue J, Chen BJ, Wang QW, Shi MK, Xie XJ, Zhang L.Two middle-age-onset hemochromatosis patients with heterozygous mutations in the hemojuvelin gene in a Chinese family.Int J Hematol, 2014, 99(4): 487–492.
猜你喜欢
中国现代医生(2022年19期)2022-11-04 10:13:29
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
趣味(作文与阅读)(2021年9期)2022-01-19 01:26:10
基层中医药(2021年8期)2021-11-02 06:24:54
种子(2021年3期)2021-04-12 01:42:22
现代临床医学(2019年4期)2019-09-10 07:43:56
Coco薇(2017年7期)2017-07-21 16:42:29
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
西南军医(2015年1期)2015-01-22 09:08:34
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
