
木质纤维素酶基因资源挖掘及真菌酶系改造
2014-05-04 08:05:10魏勇军周志华
生物加工过程 2014年1期
邹 根,刘 睿,魏勇军,周志华,严 兴
(中国科学院 上海生命科学研究院 植物生理生态研究所 合成生物学重点实验室,上海 200032)
木质纤维素是地球上蕴藏太阳能量最大的部分,具有可再生和储量丰富等优点,仅把每年再生的木质纤维素折合成能量就相当于目前人类每年消耗化石能量的20倍[1]。充分利用木质纤维素作为工农业生产的原材料是我国经济社会可持续发展的必然选择。木质纤维素必须经过不同程度的水解后才能转化为农业(动物饲料)、工业(溶剂及其他化学品,造纸和纺织原料)及能源(燃料乙醇)等行业所需的生产原料。采用生物酶系降解木质纤维素来取代原有酸碱水解的方法,不仅可以提高木质纤维素原料的利用率,而且可以极大地减少对环境的污染[2]。因此,如何构建高效的纤维素降解酶系以适应不同行业的需求,是未来发展绿色生产工艺所需的核心技术。
工业上木质纤维素酶主要由丝状真菌生产,但是其酶系存在种类不均衡、最适pH偏酸性、耐热性较差等问题,限制了其应用范围和效果。例如,在燃料乙醇和丁醇的制造过程中,工艺要求将木质纤维素中的纤维素和半纤维素都尽可能地降解为容易发酵的单糖,但常用的里氏木霉(Trichoderma reesei)的自身酶系(主要以外切酶为主)将纤维二糖转化为单糖的速度很慢,所以往往需要加入β-葡萄糖苷酶,以加快单糖的转化。又如,在造纸工艺中,要求降解木质纤维素中的木质素和半纤维素而保留其中的纤维素,因此要求丝状真菌的酶系具有比较低的内切葡聚糖酶活性和比较高的半纤维素酶活性。由于不同工艺对于酶系的耐热性和pH特性有不同的要求,并且不同种类和来源的原材料在木质纤维素的组成上差别很大,因此要求对酶系的组成作相应调整。为了适应不同原料和不同工艺的需求,经常要对工业菌株的酶系进行改造,复配甚至重构。而实现工业菌株酶系的改造,首先要了解高效木质纤维素酶系的一般规律;其次,需要收集丰富的木质纤维素酶基因资源作为候选基因;最后,如何在丝状真菌中实现外源基因的高效表达并精确调控酶系中不同成分的丰度也是酶系改造、复配和重构亟须解决的关键技术问题[3]。
1 自然界木质纤维素高效转化微生物系统的酶系组成解析
木质纤维素由纤维素,半纤维素和木质素组成,具有组成成分复杂、异质、结构高度聚合的特性。纤维素是D-葡萄糖以β-1,4-糖苷键组成的长链,在这些长链之间通过氢键与范德华力等作用形成高度有序的结晶聚合物(纤维素束)[4-5]。半纤维素的主链主要由木糖和甘露糖构成,侧链由阿拉伯糖和半乳糖等单糖和很多非糖基团构成[4-5]。木质素则是含有氧代苯丙醇或其衍生物结构单元的芳香性高聚物。虽然木质纤维素的结构复杂,一般情况下在自然界中很难被降解,但是自然界也存在着非常高效的纤维素降解微生菌(如Clostridium thermocellum和T.reesei等)或者微生物群落(如白蚁肠道微生物群落等)。对这些高效的微生物系统进行解析,了解其中的木质纤维素降解机制,将会为工业菌株的酶系改造、复配甚至重构提供坚实的理论基础和指导。
真菌及放线菌的纤维素酶游离酶系统和厌氧微生物的纤维小体系统被认为是微生物所常用的木质纤维素降解机制。但在一些高效纤维素降解菌中(如Caldicellulosiruptor saccharolyticus),既没有发现纤维小体相关结构,也没有发现游离酶系统的外切纤维素酶,这说明微生物中还存在其他的高效木质纤维素降解机制[6-7]。近些年,随着研究工作的深入,人们对于木质纤维素降解机制的多样性有了更多的认识。
在细菌研究方面,通过对C.saccharolyticus DSM 8903和同属其他7株菌(具有不同的纤维素降解能力)的泛基因组分析,确定一个含有GH9、GH48和CBM3结构域的纤维素酶对于菌株的高纤维素降解能力起关键作用[8]。敲除纤维素降解菌Clostridium phytofermentans ISDg一个属于GH9家族的纤维素酶Cphy3367(基因)之后,该菌就不能在以纤维素为唯一C源的培养基上生长,这说明该纤维素酶在C.phytofermentans ISDg的酶系中起到了关键作用[9]。
在丝状真菌研究方面,通过对里氏木霉的工业菌株Rut C30和其野生菌株的基因组学、转录组学和蛋白质组学的比较研究[10-13],深入阐述工业菌株高产纤维素酶的机制。首先,工业菌株中碳代谢阻遏因子Cre1的部分缺失使其木质纤维素酶合成不再受到葡萄糖的阻遏。同时,该菌株还缺失了85 kb的大片段,其中包含和分泌途径中糖基化修饰有关的蛋白等,这些是工业菌株能够高效分泌纤维素酶的原因之一。此外,还发现里氏木霉在纤维素诱导下高表达的酶并局限于纤维素酶和半纤维素酶,如Swollenin、Cip1和Cip2等辅助蛋白都是高表达的蛋白,表明这些辅助蛋白在木质纤维素降解过程中也起着重要作用。草酸青霉是由山东大学曲音波教授分离到的一株纤维素酶高产菌株,经过长期的诱变获得了产酶能力进一步提高的突变株Ju-A10-T。笔者所在课题组和曲音波教授课题组合作,对草酸青霉(Penicillium oxalicum)的野生株(114-2)和突变株(JU-A10-T)进行基因组、分泌蛋白组、转录组等多方面研究,揭示了草酸青霉的酶系特点和突变株高产纤维素酶的原因。草酸青霉比里氏木霉有着更加完善的酶系,例如,其β-葡萄糖苷酶不仅酶活高而且耐热性好,有良好的工业应用前景。而突变株与野生株相比,在纤维素和麸皮诱导条件下分泌的酶系更加优化,野生株114-2中较多的蛋白酶和淀粉酶在突变株JU-A10-T中的表达量明显下降,而木质纤维素酶比例则上升(图1)。进一步研究发现,突变株酶系的优化除了和一些转录因子的突变有关外,还和一些纤维素酶本身的启动子突变有关,这些研究无疑给进一步优化木质纤维素酶系提供了极为有意义的指导作用[14-17]。
在微生物群落的研究方面,Pope等[18]在对袋鼠肠道微生物群落进行元基因组学分析后,发现了一个新的木质纤维素酶降解系统,其中负责水解产物(寡糖)转运的基因(susC和susD)与淀粉水解产物转运系统非常相近。中国科学院微生物研究所东秀珠教授在牦牛瘤胃的元基因组研究中也发现了类似的木质纤维素降解机制[19]。笔者所在课题组对培菌白蚁的肠道微生物进行了元基因组测序,揭示了白蚁、真菌和白蚁肠道微生物之间的共生关系,发现肠道微生物在木质纤维素降解过程中也起到了重要的作用(水解半纤维素的支链和纤维寡糖)[20]。
随着对木质纤维素降解机制研究的深入,人们已经认识到木质纤维素的降解不仅需要纤维素酶(内切葡聚糖酶、外切葡聚糖酶和β-葡萄糖苷酶)和种类多样的半纤维素酶(糖基水解酶及糖酯酶),还需要一些氧化还原酶等辅助蛋白的参与[21]。例如,糖基水解酶GH61家族,最初被认为是一种糖苷水解酶,但最新的研究表明它实际上是一类铜离子依赖的多糖单加氧酶[22-23],它通过氧化还原反应催化纤维素结晶区的葡聚糖链断裂,形成氧化和非氧化纤维糊精,提高纤维素的水解效率。朱红秘孔菌(Pycnoporus cinnabarinus)来源的纤维二糖脱氢酶主要通过氧化还原反应断裂纤维素链,产生氧化和非氧化纤维糊精,为纤维素酶提供更多反应末端,使得里氏木霉分泌酶系对麦秆的降解效率大幅提高[24]。里氏木霉中发现的Swollenin,可以破坏葡聚糖链间的氢键,使大块纤维素聚团发生松团并有明显解聚现象,造成木质纤维素结晶度下降从而提高纤维素酶的吸附能力[25-26]。充分利用这些木质纤维素降解相关的辅助因子,将会极大提高对木质纤维素的利用效率。
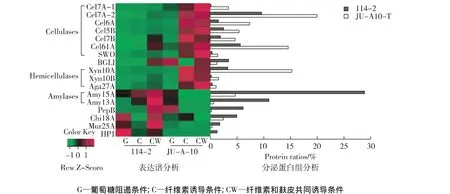
图1 草酸青霉主要胞外蛋白的表达情况和组成比例Fig.1 Expression levels and composition of main extracellular proteins of Penicillium oxalicum
2 木质纤维素酶的分类和CAZy数据库
很多序列差异很大的糖基水解酶可能催化同样的酶反应,同时同一种酶又往往能够催化多种不同类型的酶反应,所以基于酶的催化反应类型和作用底物的EC分类方法在糖基水解酶的分类上具有明显的缺陷。人们后来还发现,即使三维结构相似的糖基水解酶也会具有完全不同的催化特性,并且很多其他的木质纤维素酶也有类似的特点。由于木质纤维素酶的结构与功能之间存在复杂的对应关系,所以建立一个能够体现结构和功能关系的分类系统,是实现木质纤维素酶基因资源高效挖掘和利用的关键。
1991年法国科学家Bernard Henrissat提出按照氨基酸序列的相似性来对糖基水解酶进行分类的方法。随后,他将类似的方法扩展到了其他参与糖及糖基复合物的合成、降解和修饰相关酶的分类上,并于1998年正式发布了在线的CAZy数据库,其中包括糖基水解酶(GH)、糖酯酶(CE)、糖裂解酶(PL)和糖基转移酶(GT)(统称为碳水化合物活性酶,carbohydrate-active enzymes,CAZyme)以及碳水化合物结合结构域(CBM)[27]。2013年,CAZy数据库把木质纤维素降解所需的辅助蛋白(auxiliary activities,AAs)也收录进来,其中主要包括了木质纤维素降解所需的氧化还原酶[28]。目前,CAZy数据库已成为注释碳水化合物活性酶并进行功能预测的重要依据,几乎所有分析微生物基因组与元基因组的研究工作都会参考此数据库[29]。随着基因组和元基因组测序的开展,CAZy数据库已收集糖基水解酶序列超过13万余条(分为131个家族,但其中5个家族已经被取消),多糖裂解酶序列超过3 000条(分为22个家族),糖酯酶序列超过1万条(分为16个家族)[27]。其中有49个糖基水解酶家族、10个糖酯酶家族及3个多糖裂解酶家族与木质纤维素降解相关。
随着序列数量的增多,基于CAZy数据库的分类系统也遇到了一些问题和挑战。首先,新增的序列中大多数都没有进行酶活性分析。到2007年底,只有不到10% 的序列做过酶学活性分析,现在这一比例还在不断降低,这导致CAZy数据库在序列功能预测和注释方面越来越不可靠[27]。其次,随着序列数量的增多,CAZy数据库分类已经显得越来越粗糙,通常仅包括了clan和family2个基本的层次,只有少数家族进行了subfamily划分,这样粗层次的分类造成了同一家族内包含了多种不同活性和底物特异性的酶,不一致的功能信息还会影响新序列的 功 能 预 测[30-31]。 针 对 以 上 问 题,Bernard Henrissat团队对CAZy数据库中的GH5、13和30家族做了进一步的亚家族分类。以与木质纤维素的降解密切相关的GH5家族为例,它是整个CAZy中最大的家族之一,通过亚家族分类,其80%的基因被划分到51个不同的亚家族中。有31个亚家族做过酶学分析,其中的17个亚家族都只对应了一种酶活性,剩余的14个亚家族虽然对应2种以上的酶活性,但是往往以一种酶活性为主[30]。GH5家族的亚家族划分也与其来源菌种所处的系统发育地位一致。这些结果说明:对CAZy数据库收录基因做进一步的整理与系统分类,可以更好地反映基因序列结构与其功能、来源物种和系统发育地位之间的相关性,而一个优良的分类系统对于木质纤维素酶基因资源的挖掘和应用是非常关键的。
由于需要进行大量的序列精确比对以及应用多种方法来构建可靠的系统进化树,所以亚家族的划分费时费力,这也是相关工作进展缓慢的重要原因。最近,Busk等[32]提出了一种肽段模式识别(peptide pattern recognition)算法来进行亚家族的划分。这种方法首先需要建立每个亚家族的保守肽段库,然后根据糖基水解酶所具有的保守肽段的数量来确定亚家族的归属。因为不用进行大量的序列比对和建树的工作,所以这是一种简单快速的方法,而且也获得了很好的分类结果[32]。相信这种方法在将来不仅会用于糖基水解酶的分类,而且也有可能应用于其他的木质纤维素酶的分类。
木质纤维素酶往往都具有多个结构域,以上所述的分类方法,基本上都只考察了催化结构域的序列,而对非催化结构域则基本不予考虑。最近的研究表明,非催化结构域对于酶的功能也有很明显的作用[33],所以非催化结构域在木质纤维素酶的精细分类方面可能也会起到一定的作用。
3 木质纤维素酶基因资源的挖掘
3.1 通过基因组和元基因的序列注释
随着测序技术的进步,已经有越来越多的微生物基因组、转录组和元基因组被测序。目前,已经有23 910个细菌基因组、649个古菌基因组和5 818个真核生物基因组或转录组已经完成测序或正在测 序[34-35],其 中 包 括 了 C.thermocellum ATCC 27405、Acidothermus cellulolyticus 11B、Caldicellulosiruptor saccharolyticus DSM 8903、C.phytofermentans ISDg 和丝状真菌 T.reesei、P.oxalicum、Myceliophthora thermophila、Thielavia terrestris等高效降解木质纤维素的微生物[36]。Medie等[37]发现大约 40%的细菌基因组中至少含有1个纤维素酶基因,而在纤维素降解菌的基因组中木质纤维素酶基因的数量更高。这些基因组序列是获取新的木质纤维素酶基因资源的重要来源。
在元基因组测序方面,目前已完成396个测序项目(包含3 070个试样),包括了瘤胃、白蚁肠道、厌氧沼气发酵系统等许多木质纤维素高效降解系统的元基因组数据[35]。Hess等[38]对牛瘤胃中柳枝稷上黏附的细菌进行了元基因组测序分析,共发现了27 755个新的碳水化合物活性酶基因(主要是木质纤维素降解基因)。这些获得的新基因极大地扩展了碳水化合物活性酶基因的数量,同时也表明元基因组是获取新的木质纤维素酶基因资源的重要来源[38]。但是由于元基因测序大多数使用二代高通量测序技术(454、Solexsa或者SOLiD),序列较短且多数未能拼接,所以大部分都是不完整的基因序列。随着测序技术的进步(读序更长、序列更多)和拼接技术的完善,可以预期通过对元基因组注释获得数量更多、质量更高的木质纤维素酶基因资源。
如何对这些大量的基因组、转录组和元基因组数据进行高通量的、准确的注释,是进行木质纤维素酶基因资源挖掘所需要解决的关键技术问题。对木质纤维素降解基因进行注释的基本方法是通过blast和hmm-search,通过找到相似序列来进行家族分类和功能预测。目前,dbCAN数据库(http:∥csbl.bmb.uga.edu/dbCAN/)可以在线对用户提交的基因组数据进行自动而全面的碳水化合物活性酶注释(涵盖了木质纤维素酶基因)[29],并且dbCAN数据库还对很多元基因组来源的木质纤维素酶进行了自动注释和收集。
由于木质纤维素酶基因序列的多样性和模块化的结构以及(元)基因组测序产生的持续增加的数据量,使得CAZy数据库在功能物种预测方面的准确性大幅下降。要确定一个新发现的木质纤维素酶基因的功能,往往需要对几个甚至十几个底物进行催化反应分析后才能确定,这严重地制约了资源的利用效率。所以,要提高木质纤维素酶基因注释的准确性和效率,则有待于在CAZy数据库分类的基础上做进一步的完善,建立一个更加能体现结构与功能关系的分类系统。
3.2 通过(元)基因组文库的功能筛选
通过(元)基因组文库的功能筛选是获得木质纤维素酶基因资源的另外一种途径,由于这种方法不依赖于已知序列的相似性,因此可以与序列注释的方法互补,理论上有可能筛选到更为新颖的木质纤维素酶基因。国内外的研究者通过对(元)基因组文库的功能筛选已经从各种环境试样中筛选到了大量新颖的木质纤维素酶基因[39]。
利用功能筛选的方法从环境试样中筛选木质纤维素酶,需要构建高质量的元基因组文库,但是由于某些环境下微生物群落的生物量特别低(例如白蚁肠道),所以很难构建出高质量的、插入大片段的元基因组文库。笔者所在课题组从2个方面对白蚁肠道微生物试样的DNA提取方法进行了优化:一方面,建立了以胰酶解离法结合差速离心的方法,有效降低了肠道微生物DNA提取过程中来自白蚁基因组DNA的污染;另外一方面,将多重置换扩增技术(multiple displacement amplification,MDA技术)应用于白蚁肠道元基因组DNA的全基因组扩增,可以在群落结构不发生明显变化的基础上获得足够的高质量元基因组 DNA[40]。这些技术方法的建立,为元基因组学方法应用于白蚁肠道微生物群落的研究打下了坚实的基础,并为其他生物量低的环境试样进行元基因组研究提供了借鉴。利用这套技术方案,笔者所在课题组成功地构建了一个培菌白蚁和一个食木白蚁的肠道微生物元基因组fosmid文库并对内切葡聚糖酶、外切葡聚糖酶、β-葡萄糖苷酶和木聚糖酶进行了功能筛选,获得了几百个木质纤维素酶阳性克隆,并克隆和表达了一些具有工业应用前景的木质纤维素酶,其中一个β-葡萄糖苷酶(Bgl-gs1)的最适温度达到90℃,在75℃保温2 h,仍然能保留70%以上的酶活性[41]。
另外,笔者所在课题组还构建了沼气发酵群落的元基因组fosmid文库,并对内切葡聚糖酶、外切葡聚糖酶、β-葡萄糖苷酶和木聚糖酶进行了功能筛选,获得了上千个阳性fosmid克隆。优化了对这些fosmid克隆进行批量测序的方案,从中获得了大量有活性的木质纤维素酶基因,将部分基因在丝状真菌Rut C30中表达后,显著优化了丝状真菌中Rut C30的酶系组成[42-43]。例如,将来自沼液元基因组文库的纤维素酶基因exo2b的催化区和里氏木霉的cbh1基因通过linker融合,并用优化后的cbh1启动子 pcbh1m2[44]和 trpC 终止子组成表达盒[42]在里氏木霉中表达后,和出发株相比,转化子的发酵液水解预处理玉米秸秆所产生的葡萄糖增加了19.8%(图 2)[42,44]。
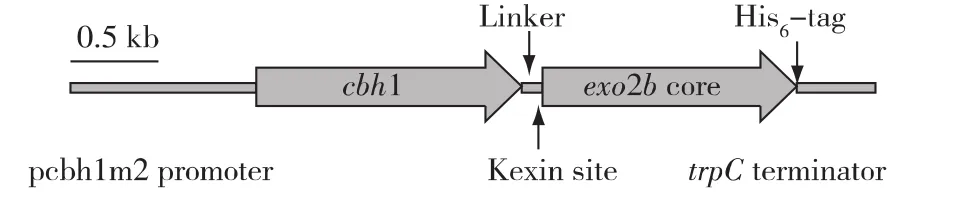
图2 从沼气厌氧发酵群落筛选到的内切葡聚糖酶基因exo2b和里氏木霉自身cbh1基因融合表达示意Fig.2 Schematic structure of exo2b(an endoglucanase screened from the metagenomic library of biogas digester)fused with a Trichoderma reesei endogenous gene cbh1
因为(元)基因组文库的构建往往是在大肠杆菌中进行的,所以目前功能筛选往往是以大肠杆菌为宿主来进行的。但是由于大肠杆菌存在如密码子偏好性和调控元件的缺失等问题,功能筛选不一定筛选到所有的具有相应功能的基因。据Gabor等[45]的研究,元基因组文库中的基因大约只有40%可以在大肠杆菌中表达,据此估计很多的木质纤维素酶基因在以大肠杆菌为宿主大肠杆菌中并没有表现出功能。为了解决这一问题,可以将一些宽宿主的复制元件(例如RK2载体的复制元件)加入文库构建载体中,这样构建好的元基因组文库就可以从大肠杆菌中转移到其他宿主(包括多数革兰氏阴性菌和少数革兰氏阳性菌)中进行功能筛选[46]。
4 丝状真菌中木质纤维素降解基因的异源表达
因为丝状真菌具有丰富的木质纤维素酶酶系,且胞外蛋白的分泌水平相比其他微生物具有很大的优势,所以用于工业化生产的纤维素酶大多来自于真菌,比较典型的生产菌株有木霉属(Trichoderma)[13]、曲霉属(Aspergillus)[47]和青霉属(Penicillium)[14,17]等。用于纤维素酶生产的丝状真菌往往经过了长期的诱变,蛋白分泌量达到了非常高的水平,例如里氏木霉Rut C30胞外蛋白水平可达 100 g/L 以上[14,48]。
由于这些丝状真菌自身的酶系往往不能满足不同工艺和原材料的要求,所以需要对酶系做进一步的完善。近年来随着丝状真菌遗传操作系统的不断完善,通过表达外源的木质纤维素酶基因,可以弥补丝状真菌自身酶系的不足[47]。但由于木质纤维素酶来源丰富多样,其中一些异源蛋白在真菌表达系统存在表达量较低或者难以表达的情况。为了解决这个难题,研究人员进行了多方面的尝试,并取得了很多有益经验,主要集中在表达宿主、表达载体、外源基因优化等几方面。
在宿主的改造方面,主要基于早期一些通过传统诱变方法选育出的优良菌株作进一步优化[10]。特别是这些优良菌株基因组的解析,对表达宿主的定向进化提供了很大的帮助。笔者所在课题组和山东大学曲音波教授实验室合作对草酸青霉基因组、转录组和分泌蛋白组解析发现,转录调控因子(CreA、AmyR等)的相关突变是菌株纤维素酶活提高的关键所在[15-17]。因此,根据这些研究发现的调控因子,对表达宿主进行遗传改造后来表达异源蛋白,可以获得较高的产量[11]。此外,还可以利用全局性转录激活因子(如Xyr1)的敲除来构建无背景的表达宿主,再通过组成型表达启动子可以获得较纯的目标纤维素酶[49-50]。宿主分泌途径中一些蛋白酶体会降解不能够正确折叠的异源蛋白,对这些蛋白酶体进行敲除可以提高异源蛋白的分泌量,同时该途径中的Pdi等分子伴侣的过表达也可以提高蛋白的表达量[51];异源蛋白分泌到胞外以后,还有可能被胞外蛋白酶降解,也有研究者通过敲除胞外蛋白酶来提高异源蛋白的成功率[47,52]。笔者所在课题组通过对里氏木霉和毕赤酵母的分泌途径进行研究发现,木霉分泌途径的糖基化水平更加适合真菌来源的BglS的表达,可以获得更高的活性和稳定性[53],这和Rut C30在诱变过程中导致的分泌能力增强有关。木霉中独特的ENGase endo T起了关键作用,它不仅可以用于其他容易过糖基化的表达系统的改造,也可以进一步调控其表达量,将木霉改造成适合不同糖基化程度的表达宿主,这些都说明里氏木霉作为一个表达宿主具有很大的应用前景。
表达载体方面最主要是启动子的优化。微生物表达系统中的启动子主要分为组成型启动子和诱导型启动子。丝状真菌表达系统组成型启动子常用的有 gpdA、trpC、pdc、pki等强组成型启动子[49-50],一般用于不需要诱导的发酵条件。诱导型启动子,在某些诱导条件下比gpdA等组成型启动子效果要好很多,而木质纤维素酶的发酵本身就是诱导条件,因此选用如cbh1等强诱导型启动子是表达纤维素酶的首选[54]。但是这些启动子上的很多诱导和阻遏因子(Xyr1、Cre1、ACE1等)的结合位点影响着外源基因的转录水平,笔者曾通过在cbh1启动子上用激活因子ACE2和Hap2/3/5蛋白复合体的结合位点取代阻遏因子Cre1的结合位点成功提高了启动子的活性[44]。
制约外源基因表达最关键的因素是基因本身。基因转录、翻译、再到分泌,均存在一些内源基因不会遇到的问题。首先,不同生物对各种密码子的使用频率不同,丝状真菌使用频率高的密码子可能在原核细胞中被用得很少。因此必须根据宿主密码子改造基因的编码序列,才能得到高表达的蛋白。此外,真核生物基因含有内含子,因此真核表达系统在表达外源基因时可能会存在由于错误剪切而导致mRNA不稳定的情况,目前已能根据宿主预测酶切位点再利用人工合成DNA或基因的定点突变来解决问题[34]。通过对序列优化外源基因能够翻译成蛋白,但是真核表达系统还需进一步通过分泌途径才能成功将蛋白分泌到胞外,分泌途径中会对一些错误折叠蛋白进行标记,然后进入解内质网相关的蛋白质降解(endoplasmic reticulum-associated degradation,ERAD),而异源蛋白往往会被标志而进入到ERAD途径。笔者所在课题组在里氏木霉中直接表达元基因组来源的纤维素酶基因exo2b未获成功,和木霉中高表达的cbh1基因通过linker融合后可以成功表达,这可能是因为内源蛋白作为Carrier可以成功通过分泌途径防止异源蛋白被识别进入ERAD[42]。上述这些方法并不是对所有的异源木质纤维素降解基因都有效。因此,如何进一步提高异源木质纤维素降解基因表达的成功率和表达量,是实现木质纤维素酶基因资源高效利用和酶系改造中亟待解决的关键技术问题之一。
5 真菌木质纤维素酶的复配和重构
在工业应用中,研究人员尝试将不同微生物来源的酶进行复配来优化构建“鸡尾酒”酶系,许多复合酶制剂都是在里氏木霉分泌酶系的基础上复配了其他微生物来源的木质纤维素降解酶[5,55],这是优化木质纤维素酶系的重要方法。里氏木霉酶系存在β-葡萄糖苷酶酶活低(仅为3 IU/mL)、半纤维素酶类少、热稳定性不佳、最适pH偏酸性等问题,这极大地限制木霉纤维素酶的应用[56]。因此,根据不同用途在木霉酶系的基础上复配合适的其他来源的木质纤维素酶是十分必要的。
Ma等[57]在里氏木霉发酵液基础上额外添加草酸青霉来源的β-葡萄糖苷酶(Pbgl1)处理玉米秸秆,还原糖产量提高80%,添加来自白蚁肠道的β-葡萄糖苷酶则可以使还原糖产量提高87%[41]。通过元基因组测序从牛瘤胃微生物群落中获得了4个活性较好的β-葡萄糖苷酶,将其中一个β-葡萄糖苷酶低剂量地添加到商品化木霉纤维素酶中,可使水解玉米秸秆的效率提高20%[58]。其次,半纤维素酶在木质纤维素降解中也发挥重要作用,它们通过降解半纤维素使内部的纤维素暴露出来,增加其与酶的接触面积,提高降解效率。Gao等[59]对温泉和草料堆肥试样进行菌群富集和筛选,获得了一批耐热性良好、酶活较高的半纤维素酶,其中包括木聚糖酶、β-木糖苷酶、α-葡糖醛酸酶和阿拉伯呋喃糖苷酶。在里氏木霉的纤维素酶中额外添加这些新发现的酶,可使水解木质纤维过程中的木糖产率提高近70%,同时酶系的耐热性和稳定性也获得了很大提升。近年来,越来越多研究表明木质纤维素降解过程中除了水解反应外,同样存在氧化还原反应,虽然在里氏木霉中也有多糖裂解单加氧酶,但是添加其他真菌来源的多糖裂解单加氧酶可以提高木霉酶系的水解效率[60-62]。而Swollenin、Cip1和Cip2等辅助蛋白则可以改变木质纤维素的结构,增加木质纤维素酶的作用效果[10,25]。在酶系复配的过程中,除了根据工艺和原料找到适当的候选酶,还需要进行大量的实验对酶系中不同酶的组成比例进行优化,以达到最佳的组成比例。酶系复配在商品化纤维素酶方面已经获得了应用,诺维信和杰能科利用这种“鸡尾酒”式的酶系复配方法成功弥补了真菌酶系的自身不足,而且避免了生产菌株异源表达多种基因的种种问题。目前,此方法成功应用于他们的商品纤维素酶制剂Cellic®CTec2和Accellerase1500生产中。
利用复配的方法尽管可以获得更完善的酶系,但是需要通过不同菌种发酵并根据用途进行配比,这会导致工业生产的步骤增多和成本升高。近年来,随着丝状真菌遗传操作系统的完善,在技术上已经可以做到在一个宿主中表达一个或者多个本身欠缺的木质纤维素酶或辅助酶,并且可以进一步敲除一些不需要的酶,从而重构出一个全新的酶系,这将为木质纤维素酶的酶系优化和生产带来新的技术变革。要在一个丝状真菌宿主中合成一个能够适应不同工业用途的全新酶系,其关键在于如何精确地调控酶系中不同成分的比例,这是研究者下一步努力的目标。
6 展望
木质纤维素酶在食品、纺织、医疗、能源等领域都有着广泛的应用,但是这些酶的生产菌株还有进一步优化空间来降低生产成本。今后,随着木质纤维素酶基因资源的挖掘和功能表征,丝状真菌遗传操作系统的完善以及近年发展起来的合成生物技术方法,我们将能够完全按照生产工艺和原材料的要求,对调控元件和木质纤维素酶标准元件进行编程式组装,在丝状真菌中实现精确可控的表达,从而利用丝状真菌“合成”出适合不同工业应用的酶系[63]。可以预见,不久的将来丝状真菌“合成”的木质纤维素酶系可以为人们的生活提供更加健康、环保和便捷的条件。
[1] Himmel M E,DingSY,JohnsonD K,etal.Biomass recalcitrance:engineering plants and enzymes for biofuels production[J].Science,2007,315(5813):804-807.
[2] Fang X,Shen Y,Zhao J,etal.Statusand prospectof lignocellulosic bioethanol production in China[J].Bioresour Technol,2010,101(13):4814-4819.
[3] Sanchez C.Lignocellulosic residues:biodegradation and bioconversion by fungi[J].Biotechnol Adv,2009,27(2):185-194.
[4] Rubin E M.Genomics of cellulosic biofuels[J].Nature,2008,454(7206):841-845.
[5] Lynd L R,Weimer P J,Van Zyl W H,et al.Microbial cellulose utilization:fundamentals and biotechnology[J].Microbiol Mol Biol Rev,2002,66(3):506-577.
[6] Xie G,Bruce D C,Challacombe J F,et al.Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii[J].Appl Environ Microbiol,2007,73(11):3536-3546.
[7] Qi M,Jun H S,Forsberg C W.Characterization and synergistic interactions of Fibrobacter succinogenes glycoside hydrolases[J].Appl Environ Microbiol,2007,73(19):6098-6105.
[8] Blumer-Schuette S E,Giannone R J,ZurawskiJV,et al.Caldicellulosiruptor core and pangenomes reveal determinants for noncellulosomal thermophilic deconstruction of plant biomass[J].J Bacteriol,2012,194(15):4015-4028.
[9] Tolonen A C,ChilakaA C,ChurchG M.Targetedgene inactivation in Clostridium phytofermentans shows that cellulose degradation requires the family 9 hydrolase Cphy3367[J].Mol Microbiol,2009,74(6):1300-1313.
[10] Baker S E,Le Crom S,Schackwitz W,et al.Tracking the roots of cellulase hyperproduction by the fungus Trichoderma reesei using massively parallel DNA sequencing[J].PNAS,2009,106(38):16151-16156.
[11] Kubicek C P,Mikus M,Schuster A,et al.Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina[J].Biotechnol Biofuels,2009,doi:10.1186/1754-6834-2-19.
[12] Seidl V,Gamauf C,Druzhinina I S,et al.The Hypocrea jecorina(Trichoderma reesei)hypercellulolytic mutant RUT C30 lacks a 85 kb(29 gene-encoding)region of the wild-type genome[J].BMC Genomics,2008,doi:10.1186/1471-2164-9-327.
[13] Martinez D,Berka R M,Henrissat B,et al.Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei(syn.Hypocrea jecorina)[J].Nature Biotechnol,2008,26(5):553-560.
[14] Gusakov A V.Alternatives to Trichoderma reesei in biofuel production[J].Trends Biotechnol,2011,29(9):419-425.
[15] Liu G,Qin Y,Li Z,et al.Improving lignocellulolytic enzyme production with Penicillium:from strain screening to systems biology[J].Biofuels,2013,4(5):523-534.
[16] Liu G,Zhang L,Qin Y,et al.Long-term strain improvements accumulate mutations in regulatory elements responsible for hyper-production of cellulolytic enzymes[J].Sci Rep,2013,doi:10.1038/srep01569.
[17] Liu G,Zhang L,Wei X,et al.Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens[J].PloS One,2013,8(2):e55185.
[18] Pope P,Denman S,Jones M,et al.Adaptation to herbivory by the Tammar wallaby includes bacterialand glycosidehydrolase profiles different from other herbivores[J].PNAS,2010,107(33):14793-14798.
[19] Dai X,Zhu Y,Luo Y,et al.Metagenomic insights into the fibrolytic microbiome in Yak Rumen[J].PloS One,2012,7(7):e40430.
[20] Liu N,Zhang L,Zhou H,et al.Metagenomic insights into metabolic capacities of the gut microbiota in a fungus-cultivating termite(Odontotermes yunnanensis)[J].PLoS One,2013,8(7):e69184.
[21] Wilson D B.Cellulases and biofuels[J].Curr Opin Biotechnol,2009,20(3):295-299.
[22] Langston J A,Shaghasi T,Abbate E,et al.Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61[J].Appl Environ Microbiol,2011,77(19):7007-7015.
[23] Phillips C M,Beeson IV W T,Cate J H,et al.Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa[J].ACS Chem Biol,2011,6(12):1399-1406.
[24] Bey M,Berrin J G,Poidevin L,et al.Heterologous expression of Pycnoporus cinnabarinuscellobiose dehydrogenase in Pichia pastoris and involvement in saccharification processes[J].Microb Cell Fact,2011,10(1):113-128.
[25] Jäger G,Girfoglio M,Dollo F,et al.How recombinant swollenin from Kluyveromyceslactis affects cellulosic substrates and accelerates their hydrolysis[J].Biotechnol Biofuels,2011,4(1):33-49.
[26] Wang Y,Tang R,Tao J,et al.Quantitative investigation of nonhydrolytic disruptive activity on crystalline cellulose and application to recombinantswollenin[J].ApplMicrobiol Biotechnol,2011,91(5):1353-1363.
[27] Cantarel B L,Coutinho P M,Rancurel C,et al.The carbohydrateactive enzymes database(CAZy):anexpertresourcefor glycogenomics[J].Nucleic Acids Res,2009,37(S1):233-238.
[28] Levasseur A,Drula E,Lombard V,et al.Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes[J].Biotechnol Biofuels,2013,doi:10.1186/1754-6834-6-41.
[29] Yin Y,Mao X,Yang J,et al.dbCAN:a web resource for automated carbohydrate-active enzyme annotation[J].Nucleic Acids Res,2012,40(W1):445-451.
[30] Aspeborg H,Coutinho P M,Wang Y,et al.Evolution,substrate specificity and subfamily classification of glycoside hydrolase family 5(GH5)[J].BMC Evolut Biol,2012,12(1):186-202.
[31] Naumoff D.Hierarchical classification of glycoside hydrolases[J].Biochemistry(Moscow),2011,76(6):622-635.
[32] Busk P K,Lange L.Function-based classification of carbohydrateactive enzymes by recognition of short,conserved peptide motifs[J].Appl Environ Microbiol,2013,79(11):3380-3391.
[33] Han Q,Liu N,Robinson H,et al.Biochemical characterization and crystal structure of a GH10 xylanase from termite gut bacteria reveal a novel structural feature and significance of its bacterial Ig-like domain[J].BiotechnolBioeng,2013,110(12):3093-3103.
[34] U.S.Department of Energy.Genomes OnLine Database [EB/OL].[2013-11-15].http:∥www.genomesonline.org/cgi-bin/GOLD/index.cgi.
[35] Pagani I,Liolios K,Jansson J,et al.The Genomes OnLine Database(GOLD)v.4:status of genomic and metagenomic projects and their associated metadata[J].Nucleic Acids Res,2012,40(D1):571-579.
[36] Rubin E M.Genomics of cellulosic biofuels[J].Nature,2008,454(7206):841-845.
[37] Medie F M,Davies G J,Drancourt M,et al.Genome analyses highlight the different biological roles of cellulases[J].Nature Rev Microbiol,2012,10(3):227-234.
[38] Hess M,Sczyrba A,Egan R,et al.Metagenomic discovery of biomass-degrading genes and genomes from cow rumen[J].Science,2011,331(6016):463-467.
[39] Duan C J,Feng J X.Mining metagenomes for novel cellulase genes[J].Biotechnol Lett,2010,32(12):1765-1775.
[40] Liu N,Yan X,Zhang M,et al.Microbiome of fungus-growing termites:a new reservoir for lignocellulase genes[J].Appl Environ Microbiol,2011,77(1):48-56.
[41] Wang Q,Qian C,Zhang X Z,et al.Characterization of a novel thermostable β-glucosidase from a metagenomic library of termite gut[J].Enzyme Microb Technol,2012,51(6-7):319-324.
[42] Geng A,Zou G,Yan X,et al.Expression and characterization of a novel metagenome-derived cellulase Exo2b and its application to improve cellulase activity in Trichoderma reesei[J]. Appl Microbiol Biotechnol,2012,96(4):951-962.
[43] Yan X,Geng A,Zhang J,et al.Discovery of(hemi-)cellulase genes in a metagenomic library from a biogas digester using 454 pyrosequencing[J].Appl Microbiol Biotechnol,2013,97(18):8173-8182.
[44] Zou G,Shi S,Jiang Y,et al.Construction of a cellulase hyperexpression system in Trichoderma reesei by promoter and enzyme engineering[J].Microb Cell Fact,2012,doi:10.1186/1475-2859-11-21.
[45] Gabor E M,AlkemaW B,JanssenD B.Quantifyingthe accessibility of the metagenome by random expression cloning techniques[J].Environ Microbiol,2004,6(9):879-886.
[46] Aakvik T,Degnes K F,Dahlsrud R,et al.A plasmid RK2-based broad-host-range cloning vector useful for transfer of metagenomic libraries to a variety of bacterial species[J].FEMS Microbiol Lett,2009,296(2):149-158.
[47] Lubertozzi D,Keasling J D.Developing Aspergillus as a host for heterologous expression[J].Biotechnol Adv,2009,27(1):53-75.
[48] Bouws H,Wattenberg A,Zorn H.Fungal secretomes:nature's toolbox for white biotechnology[J].Appl Microbiol Biotechnol,2008,80(3):381-388.
[49] Uzbas F,Sezerman U,Hartl L,et al.A homologous production system for Trichoderma reesei secreted proteins in a cellulase-free background[J].Appl Microbiol Biotechnol,2012,93(4):1601-1608.
[50] Li J,Wang J,Wang S,et al.Achieving efficient protein expression in Trichoderma reesei by using strong constitutive promoters[J].Microb Cell Fact,2012,doi:10.1186/1475-2859-11-84.
[51] Eriksson K K,Vago R,Calanca V,et al.EDEM contributes to maintenance of protein folding efficiency and secretory capacity[J].J Biol Chem,2004,279(43):44600-44605.
[52] te Biesebeke R,van Biezen N,de Vos W M,et al.Different control mechanisms regulate glucoamylase and protease gene transcription in Aspergillus oryzae in solid-state and submerged fermentation[J].Appl Microbiol Biotechnol,2005,67(1):75-82.
[53] Wei W,Chen L,Zou G,et al.N-glycosylation affects the proper folding,enzymatic characteristics and production of a fungal β ‐glucosidase[J].Biotechnol Bioeng,2013,110(12):3075-3084.
[54] Moralejo F J,Cardoza R E,Gutierrez S,et al.Thaumatin production in Aspergillus awamori by use of expression cassettes with strong fungal promoters and high gene dosage[J].Appl Environ Microbiol,1999,65(3):1168-1174.
[55] Duncan S,Schilling J.Carbohydrate-hydrolyzing enzyme ratios during fungal degradation of woody and non-woody lignocellulose substrates[J].Enzyme Microb Tech,2010,47(7):363-371.
[56] Peterson R,Nevalainen H.Trichoderma reesei RUT-C30:thirty years of strain improvement[J].Microbiology,2012,158(1):58-68.
[57] Ma X S,Zotter S,Kofler J,et al.Experimental generation of single photons via active multiplexing[J].Phys Rev A,2011,doi:10.1103/PhysRevA.83.043814.
[58] DelPozo M V,Fernandez-Arrojo L,Gil-Martinez J,et al.Microbial β-glucosidases from cow rumen metagenome enhance the saccharification oflignocellulose in combination with commercial cellulase cocktail[J].Biotechnol Biofuels,2012,doi:10.1186/1754-6834-5-73.
[59] Gao D,Uppugundla N,Chundawat S P,et al.Hemicellulases and auxiliary enzymes for improved conversion of lignocellulosic biomass to monosaccharides[J].Biotechnol Biofuels,2011,doi:10.1186/1754-6834-4-5.
[60] Mba Medie F,Davies G J,Drancourt M,et al.Genome analyses highlight the different biological roles of cellulases[J].Nature Rev Microbiol,2012,10(3):227-234.
[61] Vaaje-Kolstad G,Westereng B,Horn S J,et al.An oxidative enzyme boosting the enzymatic conversion ofrecalcitrant polysaccharides[J].Science,2010,330(6001):219-222.
[62] Harris P V,Welner D,McFarland K C,et al.Stimulation of lignocellulosicbiomasshydrolysis by proteinsofglycoside hydrolase family 61:structure and function of a large,enigmatic family[J].Biochemistry,2010,49(15):3305-3316.
[63] Seo S W,Yang J,Min B E,et al.Synthetic biology:tools to design microbes for the production of chemicals and fuels[J].Biotechnol Adv,2013,31(6):811-817.
猜你喜欢
生物化工(2022年4期)2022-09-20 09:18:08
中国特种设备安全(2021年7期)2022-01-19 05:07:32
湖南农业大学学报(自然科学版)(2021年3期)2021-07-02 01:34:24
食品与机械(2019年1期)2019-03-30 01:14:40
中外医疗(2015年5期)2016-01-04 03:57:57
恋爱婚姻家庭·养生版(2015年10期)2015-05-14 21:46:23
化工管理(2015年6期)2015-03-23 06:03:38
中国酿造(2014年9期)2014-03-11 20:21:06
湖南农业科学(2014年5期)2014-02-27 14:29:42
