
游离碳对SiC薄膜微观结构的影响①
2014-05-03 08:29金保宏王柏臣
固体火箭技术 2014年2期
金保宏,高 禹,王柏臣,陈 平
(1.沈阳航空航天大学,沈阳 110136;2.辽宁省先进聚合物基复合材料制备技术重点实验室,沈阳 110136)
0 引言
碳化硅材料具有低密度、高导热性、耐磨擦性、耐腐蚀性、抗氧化性和较低的热膨胀系数等优异性能[1-2];同时,还具有极强的刚度和硬度,使其非常适合精密型面的制备。SiC具有同质多型的特点,即在化学计量成分相同下具有不同的晶体结构。最常见的SiC多型是立方结构的3C-SiC、六方结构的4H-SiC和6H-SiC。化学气相层积(CVD)的SiC密度接近理论值、相组成单一,具有良好的机械强度、抗腐蚀性和优异的可抛光性能。所以,CVD SiC作为膜层材料受到越来越多的关注[3]。虽然在20世纪80年代,人们已在Si衬底上成功制备了可重复生长大面积3C-SiC薄膜,但SiC薄膜制备过程中存在的问题有待进一步改善,例如,SiC薄膜与衬底之间的晶格失配和热膨胀系数的差异,以及过高的沉积温度(1 200~1 380℃)等,都直接影响薄膜的质量。目前,一般采用在薄膜与衬底之间沉积缓冲层和单一气源来解决以上问题,如在Si衬底表面进行碳化处理,生长一层 SiC的缓冲层[4-5]。
传统的SiC薄膜沉积工艺存在温度高及沉积膜的SiC晶粒粗大、结构松散、其力学性能较低等问题。鉴于这类问题,本实验以Si(111)为衬底,采用一甲基三氯硅烷(CH3SiCl3)作先驱体原料,H2为载气,在高温度下制备SiC薄膜。通过采取调节反应气体配比的方法,实现SiC+C共沉积,促使SiC晶粒生长细晶化,结构致密,适当地降低沉积温度,制备含游离碳的SiC薄膜,以提高SiC薄膜的力学性能,增强SiC与基体的附着力强;并考察游离碳在SiC膜层中的分布,对SiC晶粒生长、薄膜微观结构以及薄膜力学性能的影响。
1 实验
1.1 原材料与仪器
采用的原料为一甲基三氯硅烷(CH3SiCl3,简称MTS),浙江合盛集团合盛化工有限公司,工业品。
北京中科科仪技术发展有限公司KYKY-2800B型扫描电子显微镜(SEM);日本电子株式会社JEOL2010高分辨透射电子显微镜(HRTEM);美国赛默飞世尔科技公司ESCALAB250型X射线光电子能谱仪;日本理学公司Rigaku-D/max-2400 X射线衍射仪;瑞士Leica公司MHT-4显微硬度仪。
1.2 反应原理
一甲基三氯硅烷(MTS)在H2气氛中,高温条件下的分解反应如下式[6]:

在MTS-H2体系制备SiC膜层的过程中,H2不仅是稀释气体,还作为催化剂影响反应速度和膜的组成。Yoshino M等[7]研究发现,SiC沉积质量与混合气体H2和MTS的初始浓度有关:
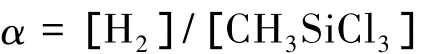
当α值过大,即H2浓度过高,导致 SiC+Si的沉积;反之,α值过小,即H2浓度过低,会导致SiC+C的沉积。在沉积过程中,气体流量和配比影响SiC膜层组成成分和表面形貌[8-9]。
1.3 实验方法
采用Si(111)单晶片作基板,并将其固定在基板支架圆盘上(石墨),在薄膜沉积腔(不锈钢)中,通入N2气排空气后,再通H2气;然后,根据设定的温度程序,升温至1 200℃,用H2气作载气,将MTS带入层积腔中,进行化学气相沉积,反应一段时间后,再改N2气保护,降温后便得到产物。
利用X射线光电子能谱仪(XPS)和X射线衍射仪(XRD)和SEM,对SiC薄膜进行成分分析和表面形貌的表征;采用TEM和HRTEM,观察和分析SiC薄膜的微观结构。
2 结果与讨论
2.1 沉积薄膜的组成成分
实验对α值分别为10和6两种条件下沉积的SiC薄膜进行考察,研究α值对SiC膜层成分的影响。SiC沉积物的XPS检测结果见图1(a)和图1(b)。对于Si-C 化合物,Si的结合能在 99.9~100.9 eV 区间,C 的结合能在281~283 eV区间。在图1(a)和图1(b)中都有Si2p峰99.9 eV 和C1s峰282.5 eV,且均在SiC 结合能的区域内。然而,对比两图的C1s峰可发现,图1(a)中只有单一的C1s峰,而图1(b)中的C1s峰出现了分裂现象,在282.5 eV峰肩上,还出现了284.6 eV的小峰,该峰位于游离态C峰结合能的284.5~285 eV区间,这表明前者为单纯的SiC膜层,而后者为SiC/C薄膜。
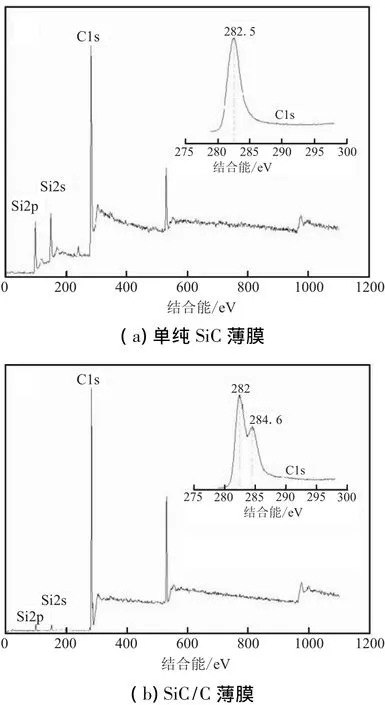
图1 SiC薄膜的XPS能谱图Fig.1 XPS patterns of SiC film
对SiC沉积物进行XRD检测,其结果见图2(a)(α=10)和图2(b)(α=6)。从图2中可知,沉积物主要为(111)面的β-SiC晶粒,还有少量的(220)、(311)及(222)等取向面的SiC晶粒。在图2(b)中还出现了石墨(002)强衍射峰,但没有石墨(004)衍射峰,这反映了游离碳的石墨化程度低;在43.4°出现了无定形碳结构的衍射峰。由此可见,沉积的游离碳为无定形碳结构。综上所述,当H2气浓度过低,会导致了SiC+C沉积,沉积的产物为SiC/C复合薄膜。
2.2 沉积薄膜的表面形貌
图3(a)和图3(b)分别为单纯SiC膜层和SiC/C膜层的SEM照片。从这2种SiC膜层的表面形貌可看出,SiC/C膜层表面较光滑,SiC晶粒细小,结构也较致密,而单纯SiC膜层的表面较粗糙,晶粒较大,结构也较疏松。这是因为在沉积SiC薄膜时,游离碳可改变SiC不同面网上的表面能,影响SiC晶粒生长速度,引起 SiC 晶粒细化[10];游离碳还会向 SiC 晶界扩散[11],填充SiC晶粒之间的缝隙,形成了富碳区,使得SiC/C膜层平整光滑,且结构致密。
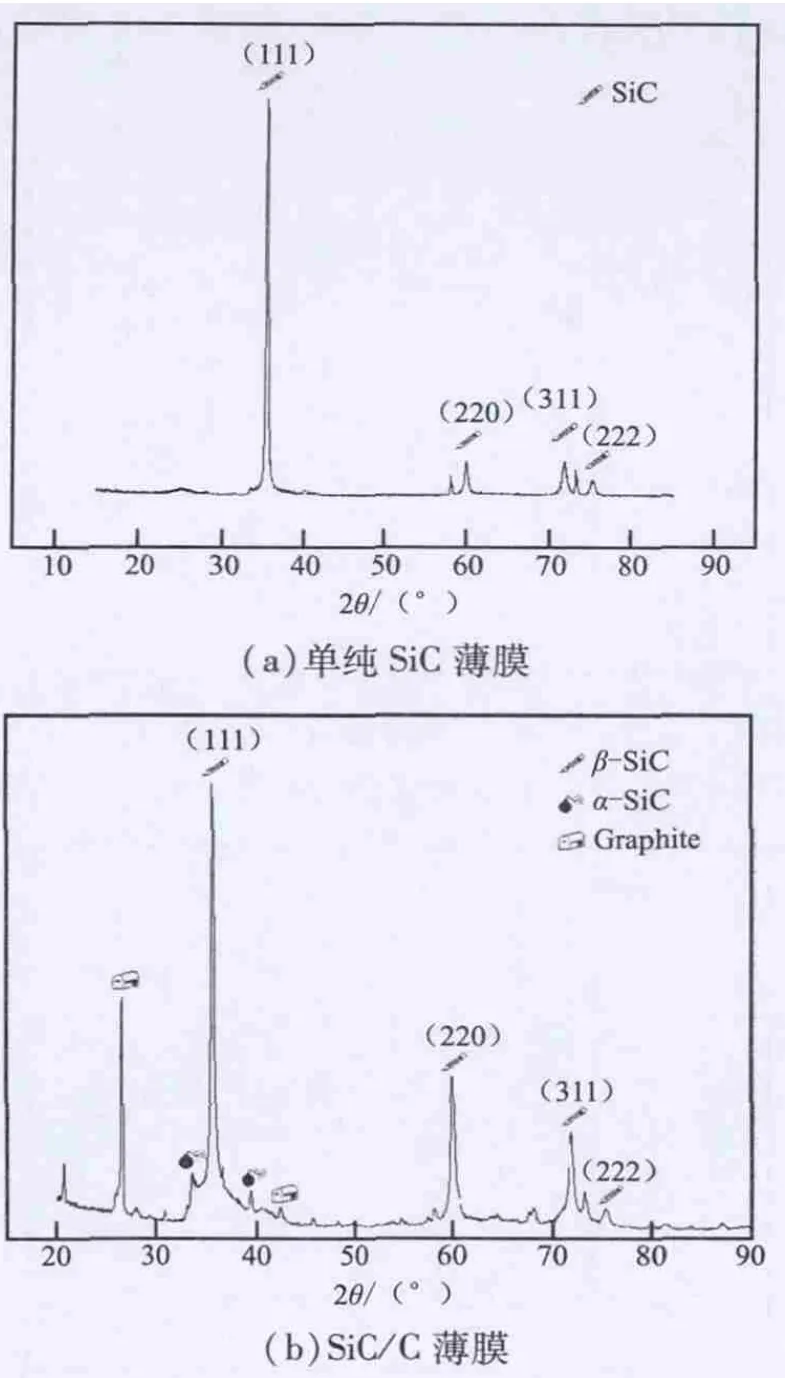
图2 SiC薄膜的X射线衍射谱Fig.2 XRD patterns of SiC film
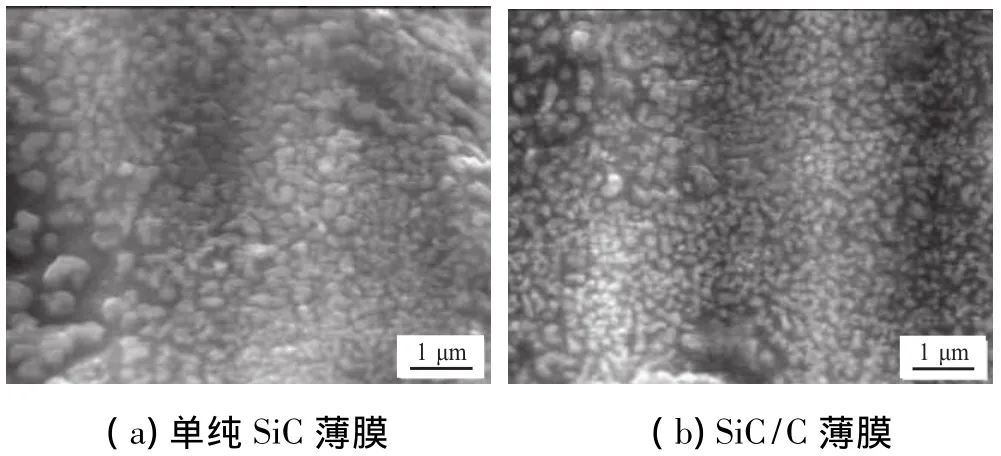
图3 SiC膜层表面的SEM照片Fig.3 SEM m icrographs of SiC film
2.3 沉积薄膜的微观结构分析
利用TEM考察SiC沉积膜中SiC的晶粒生长状况,从单纯SiC膜(图4)中可看出,SiC晶粒的轮廓清晰可见,晶粒呈柱状,为纳米级晶粒,晶粒中包含有许多衬度相间的条纹,条纹的宽度在1~3 nm之间,条带的方向垂直于晶粒的长度方向,这些衬度相间的条纹是由堆垛层错引起的[12]。图4中的插图是其选区的电子衍射花样,图像显示,SiC柱状晶沿着密排方向(111)面生长,但密排面并不是规则的堆垛,密排面方向衍射点呈线状拉长;SiC晶粒的衍射斑点点状化明显,这表明晶粒具有较高的取向度。

图4 SiC薄膜的TEM照片及其电子衍射图Fig.4 TEM m icrograph and SADP of SiC film
图5(a)和图5(b)分别为SiC/C沉积膜的TEM照片和局部的高分辨图像(HREM)。从图5(a)中可看出,SiC晶粒细小,晶粒轮廓较模糊。图5中的插图是其选区的电子衍射花样,图像显示,衍射谱呈连续的圆环状,这是SiC多晶取向随机分布的特征[13]。由于是SiC+C的沉积,所产生的游离C会覆盖到SiC晶体生长面上,干扰SiC晶粒长大。在这样的区域里,SiC晶粒重新形核或在原来晶体表面的未覆盖区域生长,其结果是形成细小的SiC晶粒;另一方面,游离碳会填充到细晶粒SiC的晶界处,阻碍晶界迁移,使晶粒合并长大变得困难[14],从而在SiC膜层中形成一个由细晶粒SiC和游离C组成的复合结构。在SiC沉积生长过程中,这样的过程被不断重复,就形成了SiC/C复合膜层。

图5 SiC/C层积膜的TEM照片及其电子衍射图Fig.5 TEM and HREM m icrographs of SiC/C film
从SiC/C复合膜层的高分辨图像(图5(b)),可进一步观察到,SiC晶粒十分细小,大部分沿密排面方向生长,呈现长和宽均只有数个纳米级的等轴晶,晶粒之间有较大的取向差,晶粒中也存在大量的层错引起的衬度条纹。同时还发现,晶粒之间存在大量非晶的碳结构,即膜层中存在富碳区,SiC晶粒与非晶的碳相互混杂在一起。
2.4 SiC薄膜的硬度测试与分析
采用MHT-4显微硬度仪,分别对α值为10和6的 SiC沉积膜进行检测,其硬度为 33.7 GPa和35.2GPa,对应的弹性模量为 257 GPa和 261 GPa。这一结果表明,实行SiC+C共沉积,适量增加碳含量,有利于提高SiC薄膜的硬度和弹性模量。这一现象可解释为由于游离碳引起SiC晶粒生长的细晶化,并填充到SiC晶界间,提高了沉积膜的光滑性和致密性,从而提高了SiC膜层的密度。此外,游离碳填充了SiC晶粒间的缺陷,在一定程度上起到钝化裂纹尖端的作用[15],极大地提高SiC/C复合膜的机械强度。当然,有关游离碳的含量对SiC薄膜的力学性能影响,还有待进一步深入研究。
3 结论
(1)采用CVD工艺制备SiC膜层的过程中,采取SiC+C共沉积时,沉积的产物为SiC/C复合膜。
(2)游离C影响了SiC晶粒生长,使得晶粒细晶化;晶粒缝隙间非晶碳的填充和形成的富碳区,有利于提高SiC/C复合膜的机械强度。
[1] Hamisch B,Kunke B.Ultra-lightweight C/SiC mirrors and structures[J].Esa.Bulletin,1998,95(8):148-152.
[2] Han JC,Zhang Y M.Optic large scale lightweight SiCmirrors[J].Journal of Astronautics,2001,22(6):124-132.
[3] Liu X Y,Zhang C R.The study of the technics on CVD SiC compact coating[J].New Technology & New Process,2002,19(12):38-41.
[4] 张荣,施洪涛.热丝法低温生长硅上单晶碳化硅薄膜[J].高技术通讯,1994,11:10.
[5] 王辉,宋航,金亿鑫.HFCVD法制备纳米晶态SiC极其室温下的光致发光[J].发光学报,2004,25(6):721.
[6] Liu Rong-Jun,Zhou Xin-Gui,Zhang Chang-Rui.SiC coating prepared by chemical vapor deposition[J].Aerospace Mater.& Tech.2002,5:42-44.
[7] Yoshino M,Shimozuma M.Deposition of SiC films by ion-enhanced plasma chemical vapor deposition using tetramethylsilane+H2[J].Thin Solid Films,2005,492:207-211.
[8] Jung-Hwan Oh,Byung-Jun Oh,Doo-Jin Choi.The effect of inputgas ratio on the growth behavior of chemical vapor deposited SiC films[J].Journal of Materials Science,2001,36(7):1695-1700.
[9] Fischman G S,Petuskey W T.Chemical vapor deposition of SiC from Si-C-Cl-H2gas system:thermochemical calculations and kinetic implication[J].Journalof the American Chemical Society,1985,68(4):185-190.
[10] 秦善.晶体学基础[M].北京:北京大学出版社,2004:148-156.
[11] 魏明坤,张广军,张丽鹏.渗硅碳化硅材料结构与性能关系的研究[J].硅酸盐学报,2002,30(2):254-257.
[12] Shinozaki S,Soto H.Microstructure of SiC prepared by chemical vapor deposition[J].J.Am Ceram Soc.,1978,61:425-426.
[13] Baohong Jin,Nanlin Shi.Analysis ofmicrostructure of silicon carbide fiber by raman spectroscopy[J].Journal ofmaterials Science & Technology,2008,24(1)261-264.
[14] 郭长友,张彩碚,贺连龙,等.游离碳对化学气相沉积SiC纤维微观结构的影响[J].金属学报,2007,43(2):165-170.
[15] Brindley P K,Draper S L,Eldridge J I,Nathal M V,Arnold SM.The effect of temperature on the deformation and fracture of SiC/Ti-24Al-11Nb[J].Metall.Trans.A.,1992,23A:2527-2540.
猜你喜欢
印染助剂(2022年11期)2023-01-03
机电安全(2022年5期)2022-12-13
实用手外科杂志(2022年2期)2022-08-31
西部林业科学(2022年3期)2022-06-29
昆明医科大学学报(2021年5期)2021-07-22
科学导报·学术(2020年29期)2020-10-21
科学(2020年1期)2020-01-06
美与时代·美术学刊(2019年9期)2019-11-29
北京航空航天大学学报(2016年6期)2016-11-16
外语教学理论与实践(2014年2期)2014-06-21
