
铜绿假单胞菌LCT-PA220菌株全基因组序列测定与分析
2014-04-21 00:45:31苏龙翔方向群刘长庭
解放军医学院学报 2014年7期
刘 超,苏龙翔,方向群,刘长庭
解放军总医院 南楼呼吸科,北京 100853
铜绿假单胞菌LCT-PA220菌株全基因组序列测定与分析
刘 超,苏龙翔,方向群,刘长庭
解放军总医院 南楼呼吸科,北京 100853
目的 全基因组测序和分析铜绿假单胞菌LCT-PA220菌株,研究空间因素对细菌全基因组的影响,为宇航员的保健奠定基础。方法通过革兰染色鉴定LCT-PA220属性,提取基因DNA、构建文库并上机测序;加工处理原始测序数据后进行基因组组装、注释和分析。结果铜绿假单胞菌LCT-PA220菌株革兰染色阴性,基因组为6.87M,包括5 829个编码序列,平均长度为962 bp,预测到为54个tRNA基因,COG数据库和KEGG数据库比对共注释到22组COG功能和32条KEGG信号通路。结论铜绿假单胞菌LCT-PA220菌株全基因组测序和基因组清楚,为后续对该菌株的研究打下了基础。
铜绿假单胞菌;基因组;测序;编码序列
铜绿假单胞菌,即绿脓杆菌,是一种非发酵革兰阴性杆菌,菌体细长且长短不一[1]。它存在于土壤、水、皮肤、呼吸道和肠道等,在自然环境中广泛分布,尤其是在潮湿的环境中,可引起人类和动物的疾病,是人类和动物的条件致病菌[2]。免疫力低下(长期使用激素类药物、免疫抑制剂、实行肿瘤放疗、化疗治疗)、侵入性诊疗操作(气管切开、保留导尿管等)或手术后的病人易受该菌感染,因此其常被认为是医院内感染的重要病原菌之一[3-4]。随着生物信息学和新一代测序技术的发展,细菌的基因组研究日趋广泛,为快速研究致病菌株的致病和耐药机制提供了有效的手段[5]。本研究分析了一株铜绿假单胞菌菌株的全基因组测序结果,为更好地了解该菌株的各种生物学特性提供了一定的理论基础。
材料方法
1 主要材料 LCT-PA220来自中国菌种保存中心CGMCC1.2387经过系列温度培养的分离株,SOAPdenovo组装软件工具(华大基因)、高通量测序仪Illumina HiSeq 2000(Illumina公司)。
2 铜绿假单胞菌LCT-PA220菌株的培养和革兰染色 取出冻存好的铜绿假单胞菌进行平板划线培养,第2天挑取单克隆菌类,接种至LB培养中30℃培养24 h。取50 μl菌液离心,弃上清,加入无菌水50 μl,吸取20 μl于载玻片上进行固定;加入革兰染色Ⅰ(结晶紫)初染1 min,自来水冲洗后加入革兰染色Ⅱ(碘液)媒染1 min,冲洗后加入革兰染色Ⅲ(酒精)脱色30 s,冲洗后加入革兰染色Ⅳ(番红)复染1 min,冲洗晾干后用油镜(100×)进行观察。
3 LCT-PA220菌株的基因组DNA提取和测序文库构建 通过裂解细菌、抽提、沉淀等步骤获得基因组DNA,采用超声将DNA打断,再将接头连接到片段上,经PCR扩增后制成测序文库,随后将已加入接头的DNA片段固定在Flow cell上,经PCR扩增反应形成Cluster。然后利用4种荧光标记的染料边合成边测序。
4 LCT-PA220菌株基因组DNA的组装和基因预测分析 原始测序数据下机后,经过过滤去除污染、重复和接头序列,得到用于组装和分析的Clean数据,运用华大自主开发的SOAP denovo组装软件对Reads数据进行组装,最终组装成全基因组序列。采用Glimmer3.0软件预测基因:通过与rRNA库比对找到rRNA,或rRNAmmer软件预测rRNA。通过tRNAscan软件预测tRNA区域和tRNA的二级结构[6-9]。
5 基因的注释和功能分析 将编码基因的蛋白质序列与各功能数据库进行比对(包括KEGG和COG数据库),得到相应基因的功能注释信息。根据每个编码序列的比对结果,为了保证其生物意义,以比对效果最好的结果为此条基因的注释。此外,还将测序得到的序列同铜绿假单胞菌的质粒数据库进行了比对。
结 果
1 LCT-PA220菌株的革兰染色 革兰染色法是细菌学研究中常用的一种鉴别染色法。由于与周围环境折光率差别异同,未经染色的细菌在显微镜下不易观察,染色后细菌与周围环境差异明显,可清晰地观察到细菌的形态、排列及某些结构特征。LCT-PA220菌株革兰染色呈现为革兰阴性杆状细菌,呈单个排列,无芽孢。见图1。
2 基因的组装和预测 使用Illumina的Genome AnalyzerⅡ进行测序并用SOAP denovo软件行组装,组装结果见表1;最终得到211个Conntigs和39个Scaffolds。G+C含量被确定为66.17%,估计菌株LCT-PA220基因组大小为6.87 M,共预测到5 829个开放阅读框,平均长度为962 bp,编码序列的长度分布见图2。tRNAscan-SE软件,预测到为54个拷贝的tRNA基因。5S、16S和23S rRNA的鉴定使用RNAmmer软件,总长度分别为114 bp、1 523 bp和2 888 bp。
3 基因功能的注释和分析 将预测后的开放读码框的蛋白序列同各数据库比对注释后,发现3 904个编码序列匹配到了22组COG的功能中,分别有2 929和3 807个CDS可分配到Swissprot和 KEGG数据库。使用铜绿假单胞菌PAO1(accession No.NC_002516.2)作为参考的基因组序列进行比较基因组分析。用SOAP比对软件分析可知所有Reads均能够比对上PAO1基因组,有大约95%的基因组覆盖率和-120X的基因组覆盖深度(图3、图4)。因此PAO1总基因长度的95%覆盖了我们的Reads。同时使用SOAPaligner发现了4个铜绿假单胞菌质粒(NC_008357,NC_009739,NC_010722和NC_007100)。但是,所有4个质粒的基因组覆盖率均<10%。来自假单胞菌的质粒CT14(accession No NC_010891)覆盖度超过31%,覆盖区域是由两个片段组成,覆盖深度为120X,表明这两个片段可能源于此质粒并整合到了该菌株的基因组中。
4 GenBank登录号 铜绿假单胞菌LCT-PA220菌株的全基因组序列存储在DDBJ/EMBL/GenBank数据库,序列号为AJKG00000000。

表1 LCT-PA220菌株基因组DNA测序数据的组装结果统计表Tab. 1 Summary of assembly of LCT-PA220 genome
讨 论
随着人类基因组计划的实施和进行,生物体基因组研究已经成为生命科学研究的前沿领域,而与之相对应的微生物基因组研究正在广泛开展[5]。细菌是微生物的一种,其基因组小、操作简单且实验周期短,其研究的工作可为人类基因组研究提供有益的参考;同时随着测序技术的更新换代和高通量测序技术的出现,细菌基因组的测序成本和所需时间已大幅度降低[10]。通过基因组研究揭示细菌的遗传规律,发现关键的功能基因并在此基础上发展疫苗,开发新型的抗菌药物或新的快速诊断方法,特别是针对某些新型传染性病原菌的全基因组测序研究,对人类的医疗卫生事业影响巨大[11]。
图 1 LCT-PA220 菌株革兰染色结果 (1 000×)Fig. 1 Gram staining of LCT-EF220 (1 000×)
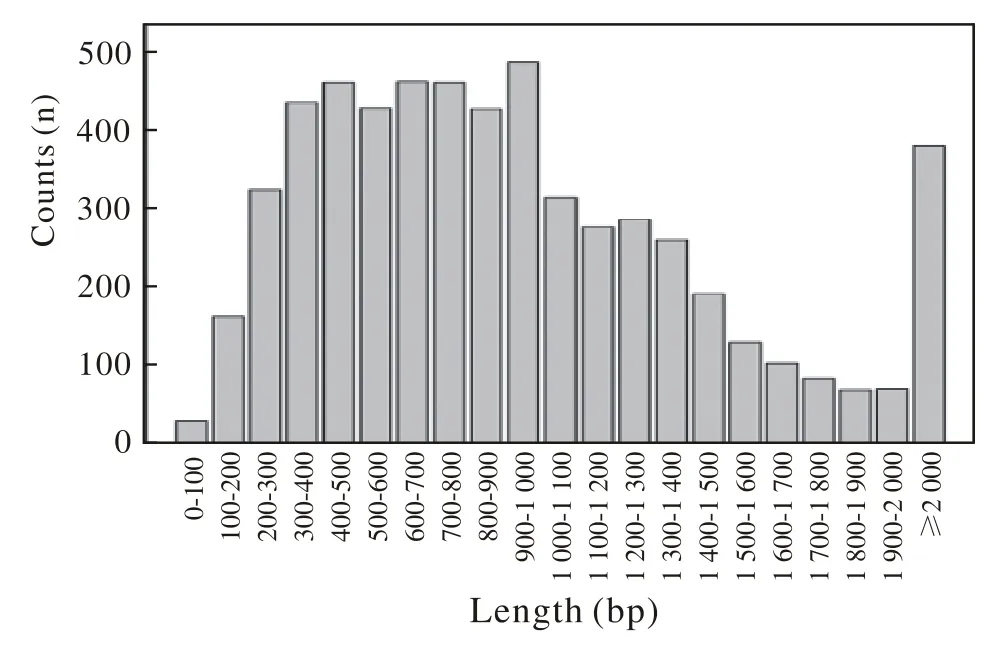
图 2 LCT-PA220菌株的基因长度分布图Fig. 2 Gene length distribution of LCT-PA220 strain
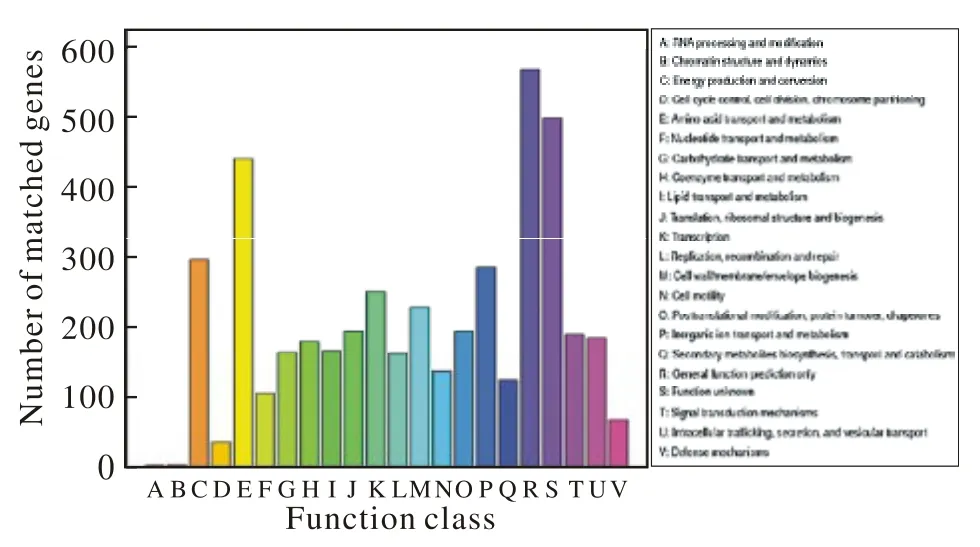
图 3 LCT-PA220菌株的COG功能分类图Fig. 3 COG function classifcation of genes in LCT-PA220 strain

图 4 LCT-PA220菌株的KEGG代谢通路分类图Fig. 4 KEGG pathway classifcation of genes in LCT-PA220 strain
铜绿假单胞菌是临床上一种常见的医院内获得性感染的机会性致病菌。其常存在于潮湿环境中,如人体呼吸道、泌尿道、消化道、伤口等处,在各种常用的医疗设备如呼吸机、导管等处也被检测到。随着有创性诊疗手段的推广和大量抗菌药物的使用,铜绿假单胞菌的感染已经上升至院内革兰阴性杆菌感染的第一位[2-3,12]。此外,国外研究尚无针对实际空间环境下的铜绿假单胞菌的全基因组测序,本研究对常见的铜绿假单胞菌衍生株LCT-PA220的全基因测序和分析,包括5 829个开放性读码框,功能基因的注释分析发现大部分编码序列都能比对上COG和KEGG数据库,KEGG代谢通路分析以感知外界环境和代谢相关的信号通路居多,COG功能分析发现大部分基因是参与生命的代谢活动,还有一部分功能未知。通过对该菌株的基因组研究,希望能揭示铜绿假单胞菌致病的遗传规律,为后续的功能基因组研究提供一些启示。
1 Hancock RE. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria[J]. Clin Infect Dis,1998, 27 Suppl 1:S93-S99.
2 Lyczak JB, Cannon CL, Pier GB. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist[J]. Microbes Infect, 2000,2(9):1051-1060.
3 Foca M, Jakob K, Whittier S, et al. Endemic Pseudomonas aeruginosa infection in a neonatal intensive care unit[J]. N Engl J Med, 2000, 343(10): 695-700.
4 Schelenz S, French G. An outbreak of multidrug-resistant Pseudomonas aeruginosa infection associated with contamination of bronchoscopes and an endoscope washer-disinfector[J]. J Hosp Infect, 2000, 46(1): 23-30.
5 Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen[J]. Nature, 2000, 406(6799): 959-964.
6 Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment[J]. Bioinformatics, 2009, 25(15): 1966-1967.
7 Delcher AL, Bratke KA, Powers EC, et al. Identifying bacterial genes and endosymbiont DNA with Glimmer[J]. Bioinformatics,2007, 23(6): 673-679.
8 Lagesen K, Hallin P, Rødland EA, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes[J]. Nucleic Acids Res,2007, 35(9): 3100-3108.
9 Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence[J]. Nucleic Acids Res, 1997, 25(5): 955-964.
10 Metzker ML. Sequencing technologies-the next generation[J]. Nat Rev Genet, 2010, 11(1): 31-46.
11 Wizemann TM, Heinrichs JH, Adamou JE, et al. Use of a whole genome approach to identify vaccine molecules affording protection against Streptococcus pneumoniae infection[J]. Infect Immun,2001, 69(3): 1593-1598.
12 Grundmann H, Kropec A, Hartung D, et al. Pseudomonas aeruginosa in a neonatal intensive care unit:reservoirs and ecology of the nosocomial pathogen[J]. J Infect Dis, 1993, 168(4): 943-947.
Whole genome sequencing and analysis of Pseudomonas aeruginosa strain LCT-PA220
LIU Chao, SU Long-xiang, FANG Xiang-qun, LIU Chang-ting
Respiratory Diseases Department in South Building, Chinese PLA General Hospital, Beijing 100853, China
LIU Chang-ting. Email:liuchangt@gmail.com
ObjectiveTo study the influence of the spatial factor which is closely associated with spacemen's health, the whole genome sequence of Pseudomonas aeruginosa strain LCT-PA220.MethodsThe characteristic was identified by gram stain examination. Then genomic DNA was extracted from LCT-PA220 strain and the library was constructed and followed by sequencing. The raw date were fltered, assembled into genome, annotated and analyzed.ResultsThe identify of LCT-PA220 strain was gram-negative bacteria whose genome was 6.87M containing 54 tRNA genes and 5 829 coding sequences with 962 bp average gene length. COG and KEGG annotation analysis revealed 22 COG functions and 32 KEGG pathways, respectively.ConclusionThe whole genome of LCT-PA220 strain is sequenced and annotated well, which paves the way for further study of this strain.
Pseudomonas aeruginosa; genome; sequencing; coding sequence
R 378
A
2095-5227(2014)07-0738-04
10.3969/j.issn.2095-5227.2014.07.025
时间:2014-03-20 15:01
http://www.cnki.net/kcms/detail/11.3275.R.20140320.1501.006.html
2014-01-16
国家“973”重点基础研究发展规划项目(2014CB744400);全军医学科研“十二五”课题重点项目(BWS12J046);武器预研重点基金(9140A26040312JB10078)
Supported by National “973” Program for Basic Science Research Development of China(2014CB744400); the Military Special-purpose Program of "Twelfth Five-Year"(BWS12J046)
刘超,男,硕士。研究方向:老年呼吸病学,微生物学。Email:c6663619@163.com
刘长庭,男,教授,主任医师,博士生导师,主任。Email:liuchangt@gmail.com
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:16:40
海洋通报(2021年1期)2021-07-23 01:55:14
现代临床医学(2021年3期)2021-07-16 07:36:22
中成药(2017年9期)2017-12-19 13:34:21
环境科技(2016年4期)2016-11-08 12:18:58
中国感染控制杂志(2015年7期)2015-12-13 08:30:42
中国感染控制杂志(2015年7期)2015-12-13 08:30:42
现代检验医学杂志(2015年1期)2015-02-06 01:59:05
中国蔬菜(2015年12期)2015-01-28 22:34:21
遗传(2014年3期)2014-02-28 20:59:09
