
SOCS超甲基化在经典型慢性骨髓增殖性肿瘤发病中的作用研究
2014-04-13 08:53:15李伶俐沈志红戎永楚康程戎奇吉梁爱斌
浙江医学 2014年16期
李伶俐 沈志红 戎永楚 康程 戎奇吉 梁爱斌
●论 著
SOCS超甲基化在经典型慢性骨髓增殖性肿瘤发病中的作用研究
李伶俐 沈志红 戎永楚 康程 戎奇吉 梁爱斌
目的 探讨细胞因子信号传导抑制蛋白(SOCS)基因超甲基化在经典型慢性骨髓增殖性肿瘤(MPN)发病机制中的作用。方法采用甲基化特异性PCR法检测220例MPN患者骨髓标本中SOCS1、SOCS3基因启动子区CpG岛甲基化发生情况,直接测序法检测MPN患者JAK2V617F、MPLW515L/K基因突变情况。应用粒细胞集落刺激因子化学诱导MPN细胞生长不同时间后,采用实时定量PCR和蛋白印迹法检测SOCS1、SOCS3 mRNA及其蛋白表达情况。同时,加入去甲基化试剂培养MPN细胞不同时间后,实时定量PCR法检测SOCS1、SOCS3 mRNA表达情况。结果MPN患者中44例(20.0%)存在SOCS1基因超甲基化,90例(40.9%)存在SOCS3基因超甲基化,156例(70.9%)JAK2V617F突变阳性,3例JAK2V617F未突变的原发性血小板增多症患者检测到MPLW515突变(其中2例为MPLW515L,1例为MPLW515K),2例JAK2V617F未突变原发性骨髓纤维化患者检测到MPLW515L突变;SOCS1、SOCS3基因超甲基化组与未甲基化组相比,其SOCS1、SOCS3 mRNA和蛋白表达量明显减少(P<0.05);JAK2V617突变组与无突变组相比,其SOCS1、SOCS3 mRNA和蛋白表达量明显减少(P<0.05);SOCS1、SOCS3基因超甲基化组,加入去甲基化试剂后SOCS1、SOCS3 mRNA表达量明显升高(P<0.05)。结论MPN患者中存在高频率的JAK2V617F基因突变和低频率的MPL基因突变以及SOCS基因启动子区CpG岛超甲基化;SOCS超甲基化和JAK2V617F突变导致SOCSmRNA和蛋白表达水平降低,异常激活JAK/STAT信号传导通路,引起细胞代谢失常而最终影响MPN的发生、发展;SOCS超甲基化是一种潜在的MPN诊断生物分子标志物及治疗靶标。
骨髓增殖性肿瘤 细胞因子信号传导抑制蛋白 JAK2基因突变 MPL基因突变 JAK/STAT信号传导通路
【Keywords】Myeloproliferative diseases Suppressor of cytokine signaling JAK2 mutation MPL mutation JAK/STAT signaling
细胞因子信号传导抑制蛋白(supperssor of cytokine signaling,SOCS)家族是一类对多种细胞因子和生长相关因子信号传导过程起负性调节作用的胞质蛋白分子,主要包括CIS和SOCS1~7等8个成员,是截至目前研究得较为透彻的JAK/STAT通路的重要反馈抑制物。SOCS1、SOCS3是SOCS家族的重要分子,可以被多种细胞因子快速诱导表达,我们选择SOCS1、SOCS3作为研究对象,探讨SOCS基因家族在经典型骨髓增殖性肿瘤(MPN)发病中的作用。
1 对象和方法
1.1 对象 选择2006-12—2012-12上海同济大学附属同济医院和慈溪市人民医院血液科门诊及住院的经典型慢性骨髓增殖性肿瘤(MPN)患者220例(上海同济大学附属同济医院130例,慈溪市人民医院90例),其中男119例,女101例,年龄18~85(51.6±10.2)岁。参照WHO分类标准进行诊断分类,其中真性红细胞增多症95例,原发性血小板增多症87例,原发性骨髓纤维化38例。根据甲基化状态及JAK2V617F突变情况将其进一步分为甲基化组与未甲基化组、突变组与无突变组。选择同期慈溪市人民医院健康志愿者50例作为对照组,其中男28例,女22例,年龄18~55(40.2±4.1)岁。MPN患者与对照组健康志愿者性别、年龄差异均无统计学意义(均P>0.05)。所有患者与健康志愿者均签署知情同意书,并经医院伦理委员会同意。
1.2 方法
1.2.1 主要试剂 DNAzol、TRIzol和M-MLV逆转录酶由美国Invitrogen公司提供。热启动Taq DNA聚合酶由德国QIAGEN公司提供。虾碱和外切酶由美国Applied Biosystems公司提供。细胞核/细胞质蛋白提取试剂盒及RNA分离试剂由北京天根生化科技有限公司提供。甲基化修饰试剂和去甲基化试剂由美国Zymo Research公司提供。化学诱导剂粒细胞集落刺激因子由深圳新鹏生物工程有限公司提供。胎牛血清及RPMI1640培养液由美国Gibco公司提供。抗人SOCS1、SOCS3单克隆抗体由美国Santa Cruz公司提供。引物和探针均由上海生工生物技术服务有限公司合成。
1.2.2 标本采集及处理 采集两组骨髓3~5ml,肝素抗凝,用Ficoll密度梯度离心法分离单个核细胞,-80℃保存待用。
1.2.3 PCR反应 (1)PCR扩增及测序:应用DNAzol试剂盒提取总DNA。JAK2V617F、MPLW515引物序列见表1。(2)PCR反应体系:80ng基因组DNA、MgCl21.25mmol/ L、dNTP0.2mmol/L、Taq酶2U、上下游引物各15pmol,总体积50μl。(3)反应条件:94℃5min;94℃30 s,60℃40s,72℃45s,35个循环;72℃10min。PCR产物经1.5%琼脂糖凝胶电泳鉴定。PCR产物采用虾碱酶和外切酶纯化,纯化产物采用ABI3700测序仪测序。
1.2.4 甲基化特异性PCR 应用DNAzol试剂盒提取DNA,与S-腺苷甲硫氨酸(终浓度为160mmol/L)、1×NE缓冲液Ⅱ和1U CpG甲基转移酶M.SSSⅠ酶混合,37℃孵育2h后再经亚硫酸氢钠修饰;引物设计见表 1。25μlPCR反应总体系含 10×PCR缓冲液 2.5μl、dNTP2.0μl、热启动 Taq酶 0.25μl,上下游引物各20pmol、模板DNA4μl,补足水至25μl。反应条件为94℃热启动11min后开始40个循环:94℃45s,60℃45s,72℃45s,最后于72℃延伸12min,4℃保存。产物经琼脂糖凝胶电泳溴乙锭(PI)染色后在紫外灯下观察,用凝胶成像系统拍照。
1.2.5 实时定量RT-PCR检测SOCS1、SOCS3mRNA水平 选取SOCS1、SOCS3甲基化MPN患者各8例(4例JAK2突变型,4例JAK2野生型),SOCS1、SOCS3未甲基化MPN患者各8例(4例JAK2突变型,4例JAK2野生型),对照组6例,抽取骨髓各5ml,经肝素抗凝,用Ficoll密度梯度离心法分离单个核细胞,将细胞分别置于盛有RPMI1640培养液的培养瓶中培养。SOCS1、SOCS3甲基化组和未甲基化组各3瓶,对照组3瓶,每瓶细胞密度≥5×106个,并加入化学诱导剂150μmol/L G-CSF,分别诱导0、1和4h。取SOCS1、SOCS3甲基化组和对照组各3瓶,加入去甲基化试剂5-氮杂脱氧胞苷(5-Aza-2′-deoxyazacytidin,5-Aza-dCR)终质量浓度为1.0g/cm3,分别反应0、24和48h。收集上述处理后的各组细胞,用TRIZOL法提取总RNA逆转录成cDNA。SOCS1探针序列为5′-FAM-CTGGAGCCAGGACCTGAACTCGCACCTAMRA-3′;SOCS3探针序列为5′-FAM-TCGCCACCTACTGAACCCTCCTCCG-TAMRA-3′;人类GAPDH探针序列为5′-FAM-GGCTGAGAACGGGAAGCTTG-TAMRA-3′。引物序列见表1。以人293细胞cDNA为模板,用SOCS1、SOCS3、HGAPDH基因引物进行PCR扩增,胶回收扩增产物,用T4DNA连接酶连接回收后产物与T载体,构建TA克隆,转化至DH5a感受态细胞,通过菌落PCR筛选阳性克隆,分析测序结果并保留测序正确的克隆。用微量紫外分光光度计Thermo NanoDrop测定质粒浓度,根据公式计算2个基因的TA克隆质粒的的基因拷贝数,制作2个基因的的标准品。PCR反应体系为25μl,含10×缓冲液2.5μl、MgCl22.0mmol/L、dNTP0.2mmol/L、上下游引物各0.5μmol/L、探针0.5μmol/L、Taq酶1.0U和cDNA50ng。反应条件:94℃5min,95℃30 s,55℃45 s,共40个循环。

表1 PCR引物序列
1.2.6 检测SOCS1、SOCS3蛋白表达水平 收集上述经G-GSF诱导0、24的48h和MPN细胞和对照细胞,采用细胞核/细胞质蛋白提取试剂盒提取细胞总蛋白,SDS-PAGE分离蛋白,半干法转膜,分别加入1∶500稀释的兔抗人SOCS1、SOCS3和兔抗GAPDH,4℃过夜,再加入1∶5 000稀释辣根过氧化物酶标记的IgG二抗,室温下孵育1~2h后用电化学发光试剂显色,最后图像扫描并分析。
1.3 统计学处理 采用SPSS15.0统计软件,计量资料以表示,组间比较采用单因素方差分析。计数资料组间比较采用χ2检验。
2 结果
2.1 JAK2V617F和MPLW515基因突变筛查结果 220例MPN患者中共有156例(70.9%)存在JAK2V617F突变(其中杂合子型149例,纯合子型7例),包括真性红细胞增多症91例(91/95,95.8%),原发性血小板增多症46例(46/87,52.9%),原发性骨髓纤维化19例(19/38,50.0%)。64例JAK2V617F无突变的患者中有5例(5/220,2.3%)检测到MPLW515突变,包括原发性血小板增多症3例(3/87,3.4%),其中MPLW515L 2例,MPLW515K 1例;原发性骨髓纤维化2例(2/38,5.3%),均为MPLW515L突变。对照组无JAK2V617F或MPLW515突变。
2.2 MPN患者JAK2突变组与无突变组SOCS1、SOCS3基因启动子区CpG岛甲基化检出情况 见表2,图1、2。
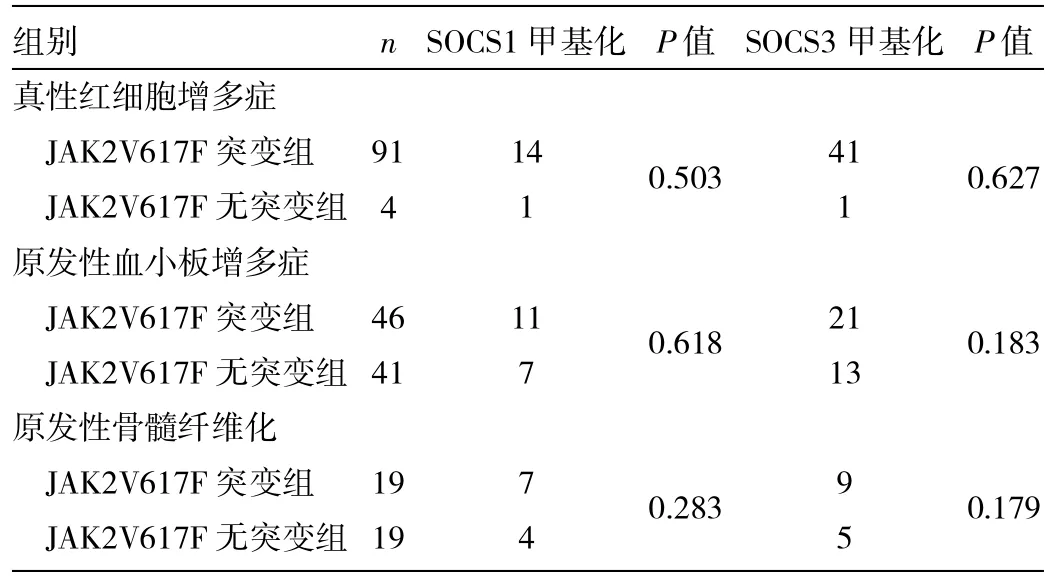
表2 MPN患者JAK2突变组与无突变组SOCS1、SOCS3基因启动子区CpG岛甲基化情况(例)
由表2可见,MPN患者中44例(20.0%)存在SOCS1基因超甲基化,包括真性红细胞增多症15例(15/ 95,15.8%),以原发性血小板增多症18例(18/87,20.7%),原发性骨髓纤维化11例(11/38,28.9%),以原发性骨髓纤维化患者频率最高,但3种病变患者的差异无统计学意义(χ2=2.980,P>0.05)。90例(40.9%)存在SOCS3基因超甲基化,包括真性红细胞增多症42例(42/95,44.2%),原发性血小板增多症34例(34/87,39.1%),原发性骨髓纤维化14例(14/38,36.8%),3种病变患者的SOCS3基因甲基化率的差异也无统计学意义(χ2=0.809,P>0.05)。MPN患者中,JAK2V617F突变组与无突变组,SOCS1、SOCS3基因甲基化率差异无统计学意义(χ2=0.088,4.702;P=0.0854,0.035>0.05)。对照组未发现有SOCS1、SOCS3基因超甲基化。
2.3 G-CSF处理不同时间后MPN细胞SOCS1、SOCS3 mRNA表达水平的变化 见图3。
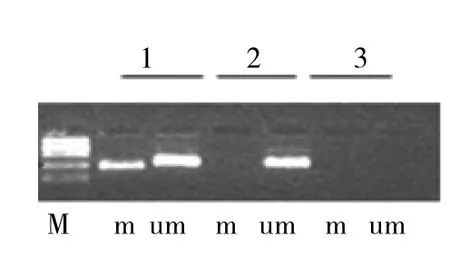
图1 SOCS1甲基化特异性PCR检测结果(1:SOCS1甲基化;2:SOCS1非甲基化;3:阴性对照;m:甲基化引物;u:非甲基化引物;M:DNA标记)
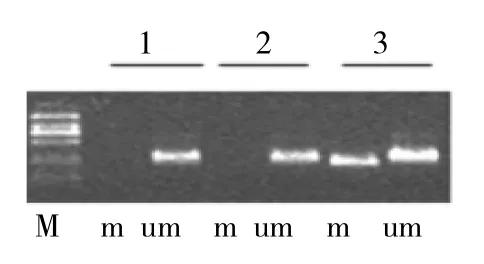
图2 SOCS3甲基化特异性PCR检测结果(1、2:SOCS3非甲基化;3:SOCS3甲基化;m:甲基化引物;u:非甲基化引物;M:DNA标记)
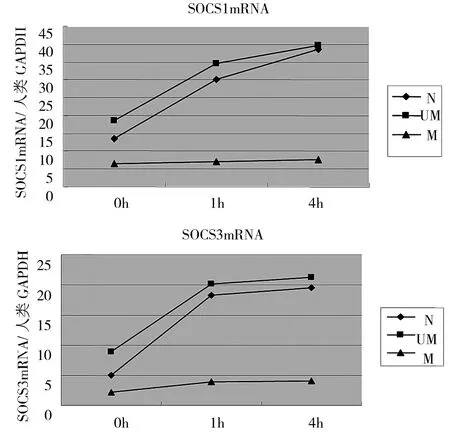
图3 G-CSF处理不同时间后MPN细胞SOCS1、SOCS3 mRNA表达水平的变化(N:对照组;UM:未甲基化组;M:甲基化组)
由图3可见,各组在G-CSF诱导0、1和4h后SOCS1、SOCS3基因表达水平升高。G-CSF诱导前,甲基化组SOCS1、SOCS3 mRNA表达平均水平低于非甲基化组和对照组(P<0.05)。经G-CSF诱导后,未甲基化组和对照组SOCS1、SOCS3 mRNA表达持续上升,甲基化组SOCS1、SOCS3 mRNA表达仍然处于低水平,显著低于非甲基化组和对照组,差异有统计学意义(P<0.05)。
2.4 G-CSF处理不同时间后MPN细胞SOCS1、SOCS3蛋白表达水平的变化 见图4。
由图4可见,G-CSF诱导前,各组SOCS1、SOCS3蛋白表达水平均较低,甲基化组低于对照组和未甲基化组,但差异无统计学意义(P>0.05)。在G-CSF诱导24h时,未甲基化组和对照组SOCS1、SOCS3蛋白表达水平均显著上升,甲基化组仍处于低水平;对照组的SOCS1蛋白表达水平48h时仍持续上升,而SOCS3蛋白表达水平24h后急剧下降;未甲基化组SOCS1、SOCS3蛋白表达水平显著高于甲基化组(P<0.05),且在G-CSF诱导下有持续升高的趋势。

图4 G-CSF处理不同时间后MPN细胞SOCS1、SOCS3蛋白表达水平的变化(N:对照组;UM:未甲基化组;M:甲基化组)
2.5 JAK2V617F突变对SOCS1、SOCS3mRNA和蛋白表达水平的影响 见图5。
由图 5可见,SOCS1、SOCS3未甲基化组中的JAK2V617F突变组SOCS1、SOCS3mRNA和蛋白表达水平均明显低于JAK2V617F无突变组(均P<0.05)。SOCS1、SOCS3甲基化组中的JAK2V617F突变组和JAK2V617F无突变组SOCS1、SOCS3mRNA和蛋白表达水平的差异均无统计学意义(均P>0.05)。
2.6 去甲基化对SOCS1、SOCS3mRNA表达水平的影响见图6。
由图6可见,在G-CSF诱导下,甲基化组加入去甲基化试剂孵育0、24和48h后SOCS1、SOCS3mRNA表达水平升高,与未加去甲基化试剂的甲基化组和对照组比较,差异均有统计学意义(均P<0.05);对照组加入去甲基化试剂0、24和48h后,SOCS1、SOCS3mRNA表达水平稍有下降,但与未加去甲基化试剂的对照组比较,差异均无统计学意义(均P>0.05)。
3 讨论
目前酪氨酸激酶JAK2突变导致JAK/STAT通路的异常激活被认为是经典型MPN发病的重要分子机制。在绝大多数的真性红细胞增多症患者、约50%的原发性血小板增多症患者和原发性骨髓纤维化患者可检测到JAK2V617F点突变[1-3],本研究结果也证实了这一结论。此后,在少数这些疾病患者中还检测到JAK2基因其他位点功能类似的点突变[4]。有研究报道在5%的原发性骨髓纤维化患者和1%的原发性血小板增多症患者检测到位于MPL跨膜区的MPLW515突变[5],该突变导致突变细胞发生细胞因子非依赖性激活,激活下游JAK/STAT途径。但仍有相当部分MPN患者无JAK2或MPL基因突变,提示还存在其它分子机制参与经典型MPN发病。

图5 JAK2V617F突变对SOCS1、SOCS3mRNA和蛋白表达水平的影响[(+)指发生突变或甲基化,(-)指无突变或甲基化]
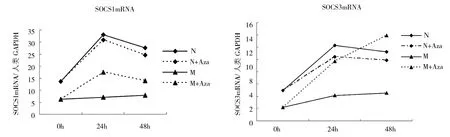
图6 去甲基化对SOCS1、SOCS3mRNA表达水平的影响(N:对照组;Aza:去甲基化试剂5-氮杂脱氧胞苷;M:甲基化组)
正常生理情况下,JAK/STAT通路介导促红细胞生成素、促血小板生成素、粒-巨噬细胞集落刺激因子等多种细胞因子的信号传导,调控细胞的增殖、分化和凋亡。SOCS蛋白家族成员作为重要的负调控因子,参与多种信号传导途径的负反馈调节,尤其在JAK/STAT通路的负反馈调节机制中起着重要作用。SOCS蛋白家族具有共同的结构:中间的SH2结构域;位于N末端的可变序列;位于C末端的由40个氨基酸残基组成的“SOCS盒”。目前研究认为,SOCS蛋白对细胞因子信号传导抑制作用主要有3种分子机制:与JAKs结合并抑制其活性;与STATs竞争结合细胞因子受体磷酸化位点,阻止STAT活化;诱导与之结合的JAKs、STATs的降解,从而阻断细胞因子的信号传递。与SOCS家族的其他成员不同,SOCS1、SOCS3还包含有一个由12个氨基酸组成的结构-激酶抑制区(KIR),可以与JAK2的酪氨酸激酶区(JH1)结合并使其失活。基因缺失实验证明不同的SOCS蛋白家族成员具有不同的高度特异性的生理功能。SOCS1、SOCS3在造血细胞的负性调控中显得尤为重要[6]。SOCS1基因缺失小鼠表现为发育障碍,且出生不到3周死亡,伴有严重的淋巴细胞减少、T淋巴细胞活化、肝细胞坏死、主要脏器的巨噬细胞浸润等特征[7-8]。Alexander等[8]进一步研究认为SOCS1在INF-γ信号传导的调控和T细胞分化中起着重要作用。Boyle等[9]发现SOCS3基因缺失小鼠最终发展为真性红细胞增多症,表明SOCS3可能在负性调控红细胞生成中起重要作用。
目前研究认为SOCS基因启动子区的超甲基化导致基因沉默可能在肿瘤的发生、发展中起重要作用[10-12]。在血液肿瘤中,如急性髓系白血病、MDS常发现SOCS基因超基化而导致基因沉默[13-14]。最近Teofili[15]等发现约1/3真性红细胞增多症和1/4原发性血小板增多症患者存在SOCS1或SOCS3超甲基化(不依赖JAK2V617F突变),导致基因转录水平降低,减弱对JAK2/STAT通路的抑制作用,对MPD的发病起了重要作用。本研究结果证实真性红细胞增多症、原发性血小板增多症患者中存在较高频率的SOCS基因启动子区CpG岛超甲基化,除此之外,我们还发现在原发性骨髓纤维化患者中也存在SOCS的超甲基化,研究显示约1/2真性红细胞增多症患者、3/5原发性血小板增多症和3/5原发性骨髓纤维化患者存在SOCS1和/或SOCS3的启动子区CpG岛的超甲基化且不依赖JAK2V617F突变。同时,本研究未发现SOCS1、SOCS3甲基化率在真性红细胞增多症、原发性血小板增多症及原发性骨髓纤维化患者中的分布差异有统计学意义。本研究结果显示SOCS1、SOCS3基因启动子区的CpG岛超甲基化导致基因表达下调,且不能通过细胞因子的刺激而升高。而去甲基化实验则进一步证明超甲基化与基因转录沉默的关系。事实上,SOCS1、SOCS3基因启动子区的CpG岛作为转录因子的结合位点,对SOCS1、SOCS3的转录激活起着重要作用。这些位点甲基化造成SOCS1、SOCS3表达量下降,不能有效行使其生理功能,对JAK/STAT通路的反馈抑制被削弱,JAK/STAT通路异常激活,导致细胞代谢失常而最终影响MPN的发生、发展。本研究还发现SOCS甲基化的频率和其转录抑制效应是一致的,当去甲基化后,SOCS1、SOCS3的转录恢复正常,可以重新反馈抑制JAK/STAT通路的活性,诱导细胞凋亡。本研究结果说明SOCS基因超甲基化可能是MPN的重要发病机制并提示SOCS1、SOCS3超甲基化可能是治疗MPN的一种潜在分子靶标。本研究结果表明约2/3的MPN患者由于SOCS基因超甲基化导致基因表达下调,在体外加入去甲基化试剂可以使其恢复表达。针对这些患者可以设计出一种分子靶向治疗方法,使沉默基因去甲基化以恢复正常转录。考虑到还有约1/3的MPN患者JAK2V617F无突变,对这些患者评估SOCS的甲基化状态显得尤为重要。
本研究还发现JAK2V617F突变对SOCS转录水平存在抑制作用。JAK2V617F突变位置恰好位于JAK2假激酶区(JH2区),破坏了该区对酪氨酸激酶区(JHl区)的抑制作用,从而发生JAK2的组成型激活。Hookham等[16]的研究表明,JAK2V617F突变的造血祖细胞对SOCS蛋白的抑制作用产生抵抗性,由于这些患者SOCS表达量无法恢复,因此抑制活化的激酶也不能恢复传导系统的诱导与抑制之间的平衡。
本研究证实了Pardanani等[5]关于原发性血小板增多症和原发性骨髓纤维化患者中存在MPLW515突变的观点。该突变的存在验证了我们先前的推测,即还有其他分子异常参与了MPN的发病。本研究中5例MPLW515突变患者JAK2V617F均未突变,这一发现为JAK2V617F未突变的经典型MPN患者的发病机制做了有益补充,但是MPLW515突变率并不高,因此对那些JAK2V617F和MPLW515均未突变的MPN患者,还有其他的分子异常参与发病。本研究结果显示SOCS基因启动子区CpG岛超甲基化可能是MPN发病的诱发因素之一,尤其在JAK2V617F未突变的MPN发病机制中可能起着重要作用。至于MPN患者中SOCS超甲基化、JAK2V617F突变及MPLW515突变3者关系如何,是否还有其他分子机制参与发病,值得进一步研究。
[1]Baxter E J,Scott L M,Campbell P J,et al.Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders[J].Lancet,2005,365(9464):1054-1061.
[2]James C,Ugo v,Le Couedic J P,et al.A unique clonal JAK2 mutation leading to constitutive signaling causes polycythaemia vera [J].Nature,2005,434(7037):1144-1148.
[3]Levine R L,Wadleigh M,Cools J,et al.Activating mutation in the tyrosine kinase JAK2 in polycythemia vera,essential thrombocythemia,and myeloid metaplasia with myelofibrosis[J].Cancer Cell,2005,7(4):387-397.
[4]Scott L M,Tong W,Levine R L,et al.JAK2 exon 12 mutations in polythemia vera and idiopathic erythrocytosis[J].N Engl J Med, 2007,356(5):459-468.[5]Pardanani A D,Levine R L,Lasho T,et al.MPL515 mutations in myeloproliferative and other myeloid disorders:a study of 1182 patients[J].Blood,2006,108(10):3472-3476.
[6]Valentino L,Pierre J.JAK/STAT signal transduction:regulators and implication in hematological malignancies[J].Biochem Pharmacol, 2006,71(6):713-721.
[7]Krebs D L,Hilton D J.SOCS:Physiological suppressors of cytokine signaling[J].J Cell Sci,2000,113(Pt16),2813-2819.
[8]Alexander W S,Starr R,Fenner J E,et al.SOCS1 is a critical inhibitor of interferonγ signaling and prevents the potentially fatal neonatal actions of this cytokine[J].Cell,1999,98(5):597-608.
[9]Boyle K,Robb L.The role of SOCS3 in modulating leukaemia inbihitory factor signaling during murine placental development[J].J Reprod Immunol,2008,77(1):1-6.
[10]Xiong H,Du W,Zhang Y J.Tricho station A,a histone deacetylase inhibitor,suppresses JAK2/STAT3 signaling via inducing the promoter-associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells[J].Mol Carcinog,2012,51(2):174-184.
[11]Hua D,Hu Y,Wu Y Y,et al.Quantitative methylation analysis of multiplegenesusing methylation-sensitive restrictionenzyme-based quantitative PCR for the detection of hepatocellular carcinoma[J].Exp Mol Pathol,2011,91(1):455-460.
[12]Sasi W,Jiang W G,Sharma A,et al.Higher expression levels of SOCS1,3,4,7 are associated with earlier tumor stage and better clinical outcome in human breast cancer[J].BMC Cancer,2010, 10:178.
[13]Watanabe D,Ezoe S,Fujinoto M,et al.Suppressor of cytokine signaling-1 gene silencing in acute myeloid leukaemia and human haematopoeitic cell lines[J].Br J Haenatol,2004,126(5):726-735.
[14]Wu S J,Yao M,Chou W C,et al.Clinical implications of SOCS1 methylation in myelodysplastic syndrome[J].Br J Haematol, 2006,135(3):317-323.
[15]Teofili L,Martini M,Cenci T,et al.Epigenetic alteration of SOCS family members is a possible patheogenetic mechanism in JAK2 wild type myeloproliferative diseases[J].Int J Cancer,2008,123 (7):1586-1592.
[16]Hookham M B,Elliott J,Suessmuth Y,et al.The myeloproliferative disorder-associated JAK2V617F mutant escape negative regulation by suppressor of cytokine signaling[J].Blood 2007,109 (11):4924-4929.
Association between SOCS gene hypermethylation and classical myeloproliferative neoplasms
Objective To investigate the association between hypermethylation of suppressor of cytokine signaling(SOCS) gene and typical myeloproliferative neoplasms(MPN).MethodsMethylation-specific PCR was used to detect CpG island methylation status of SOCS1 and SOCS3 genes in bone marrow samples from 220 MPN patients.JAK2V617F and MPLW515L/K gene mutations were detected by direct DNA sequencing assay.The expression of SOCS1 and SOCS3mRNA and protein was evaluated by real-time quantitative PCR and Western blot,after MPN cells were co-cultured with GC-SF or demethylating agent 5-aza-2′-deoxyazacytidin at different time points.ResultsThe rates of SOCS1 and SOCS3 gene hypermethylation were 20.0%(44/220)and 40.9%(90/220),respectively.JAK2V617F positive mutation was detected in 156 patients(70.9%);in 3 JAK2V617F-negative patients with essential thrombocytosis(ET),MPLW515 mutation was detected (2 MPLW515L and 1 of MPLW515K)and in 2 JAK2V617F-negative patients with idiopathic myelofibrosis(IMF),MPLW515L mutation was detected.The expression of SOCS1,SOCS3 mRNA and protein was significantly decreased in hypermethylation group compared to non-methylation group(P<0.05);and in JAK2V617F mutation group compared to wild type group(P<0.05).After co-cultured with demethylating agent the expression of SOCS1,SOCS3mRNA was increased significantly in hypermethylation group(P<0.05).ConclusionHigh-frequency JAK2V617F gene mutation,low-frequency MPL gene mutation and SOCS gene CpG island hypermethylation are associated with myeloproliferative neoplasms,and SOCS hypermethylation might be a potential diagnosticbiomarker and therapeutic target for the disease.

2013-06-09)
(本文编辑:杨丽)
宁波自然科学基金(2011A610040)
315300 慈溪市人民医院检验科(李伶俐、沈志红、戎永楚、康程、戎奇吉);上海同济大学附属同济医院血液科(梁爱斌)通信作者:李伶俐,E-mail:sztext@vip.163.com
猜你喜欢
疯狂英语·新悦读(2023年5期)2023-09-11 01:04:05
传染病信息(2022年3期)2022-07-15 08:24:28
肝博士(2021年1期)2021-03-29 02:32:16
国际放射医学核医学杂志(2020年3期)2020-07-27 01:59:16
文苑(2018年18期)2018-11-08 11:12:42
现代检验医学杂志(2016年2期)2016-11-14 02:38:26
医学研究杂志(2015年6期)2015-07-01 17:40:08
现代检验医学杂志(2015年2期)2015-02-06 02:00:48
沈阳医学院学报(2014年4期)2014-12-27 13:44:30
中国中医药现代远程教育(2014年13期)2014-03-01 04:26:57
