
Computational Investigation of the Substituent Effect on the Intramolecular Proton Transfer Reaction of 3-Hydroxytropolone
2014-03-25 02:35VedtINDilrzbkir
结构化学 2014年12期
GÜL Vedt IŞIN Dilr Özbkir
(Chemistry Department, Faculty of Science, Cumhuriyet University 58140 Sivas, Turkey)
1 INTRODUCTION
The proton transfer reaction plays very important roles in physical, chemical and biological systems[1-4].Tropolone (TRN) and its derivatives, as model compounds for intramolecular proton transfer, are explored experimentally[5-15]and theoretically[16-22].Tropolone is a seven-membered aromatic molecule having an intramolecular hydrogen bond. It can form both intramolecular and intermolecular hydrogen bonds because of its acidic OH proton and basic C=O groups. 3-OHTRN (3-hydroxytropolone, a derivative of TRN) exhibits two tautomers, 2,7-dihydroxycyclohepta-2,4,6-trien-1-one (I) and 2,3-dihydroxycyclohepta-2,4,6-trien-1-one (II)[23]. In the previous paper[24], we have reported the density functional theory (DFT) (B3LYP/6-31+G**) calculation results for the proton transfer reaction of 3-OHTRN and its dimers. In this paper, -CN, -NH2,-NO2and -CH3groups have been attached to the 3, 4,5 and 6 positions of ring in order to investigate the influence of these groups on the proton transfer reaction of 3-OHTRN (these reactions are shown schematically in Scheme 1). For this reason, we have carried out the calculations using the Becke’s three-parameter hybrid method with the Lee, Yang and Parr correlation functional (B3LYP)[25-27]in the gas phase and in solution. According to the literature data, there have been no experimental and computational studies on the proton transfer reaction between tautomers of these derivatives.
2 METHODS
The stationary structures (reactants, products and transition states) were optimized using B3LYP[25-27]method by applying at the 6-31+G** basis set with polarization functions on all atoms and diffuse functions on heavy atoms in the gas phase. The harmonic vibrational frequencies were obtained at the same level of theory used in the geometry optimizations. This allowed us to classify the stationary points found as local minima or transition states. The solute-solvent interaction was evaluated using the self-consistent reaction field (SCRF)method, which is based on the self-consistent isodensity polarized continuum model (SCI-PCM)[28].The reaction field calculation was carried out for ε =78.39 (water). All the calculations were performed with the Gaussian 03W computational package[29].
3 RESULTS AND DISCUSSION
The selected structural parameters for I, II and TS(the transition state) structures of -CN, -NH2, -CH3and -NO2derivatives of 3-OHTRN are listed in Tables 1 and 2. The atom numbering and the schematic projection are shown in Scheme 1. In addition,all reactions are shown in Figs. 1, 2, 3, and 4 (Some selected bond lengths are given on the compounds).

Scheme 1. Schematic representation of the investigated compounds
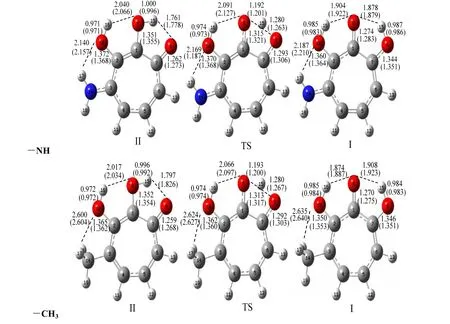
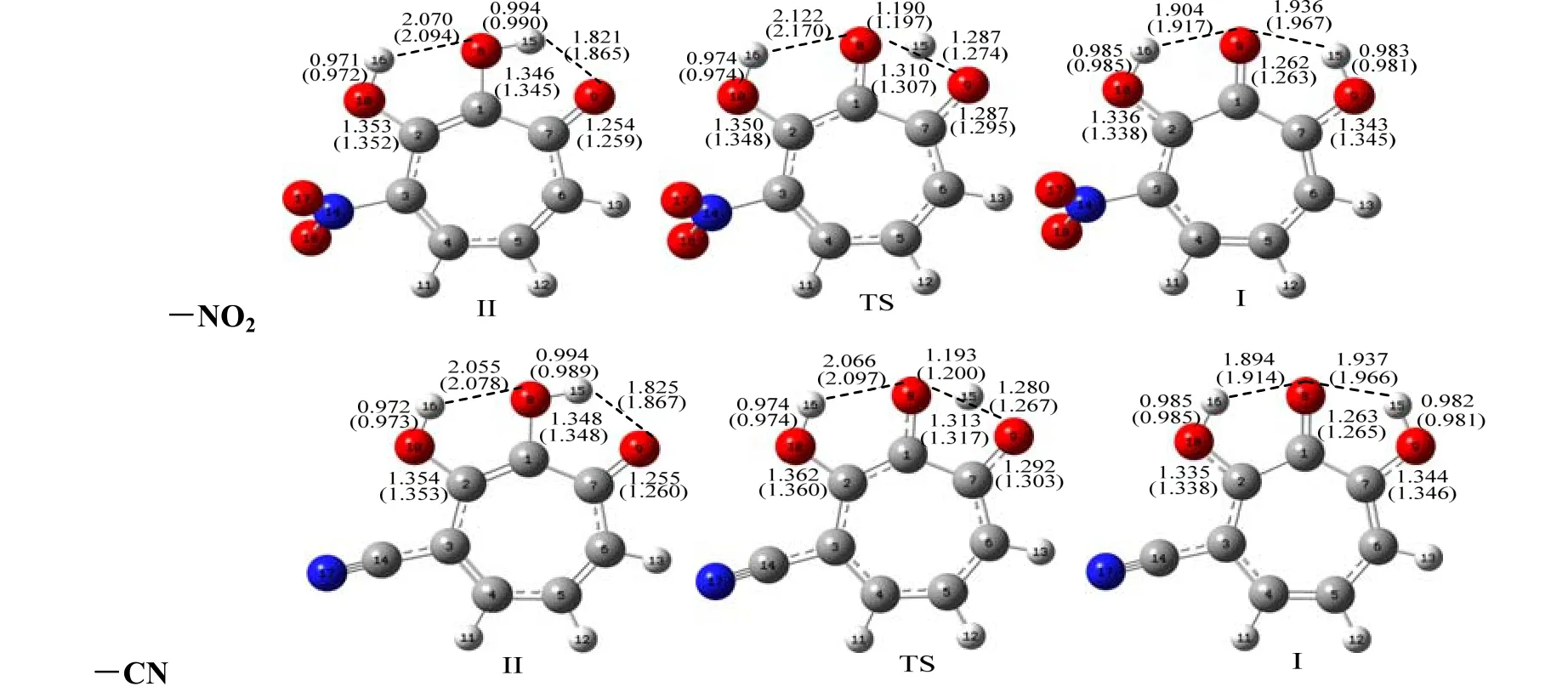
Fig. 1. Optimized structures of I, II and TS (transition state) with substituents (-NH2, -CH3, -NO2 and-CN) attached to 3 position of 3-OHTRN at the B3LYP (6-31+G**) level in the gas phase(Gauss View 3.0 [30]). Some selected bond lengths (in Å) at the same level are given on the compounds. The numbers in parentheses belong to the aqueous phase

Fig. 2. Optimized structures of I, II and TS with substituents (-NH2, -CH3, -NO2 and -CN) attached to 4 position of 3-OHTRN at the B3LYP (6-31+G **) level in the gas phase. Some selected bond lengths (in Å) at the same level are given on the compounds. The numbers in parentheses belong to the aqueous phase

Fig. 3. Optimized structures of I, II and TS with substituents (-NH2, -CH3, -NO2 and -CN) attached to 5 positions of 3-OHTRN at the B3LYP (6-31+G **) level in the gas phase. Some selected bond lengths (in Å) at the same level are given on the compounds. The numbers in parentheses belong to the aqueous phase
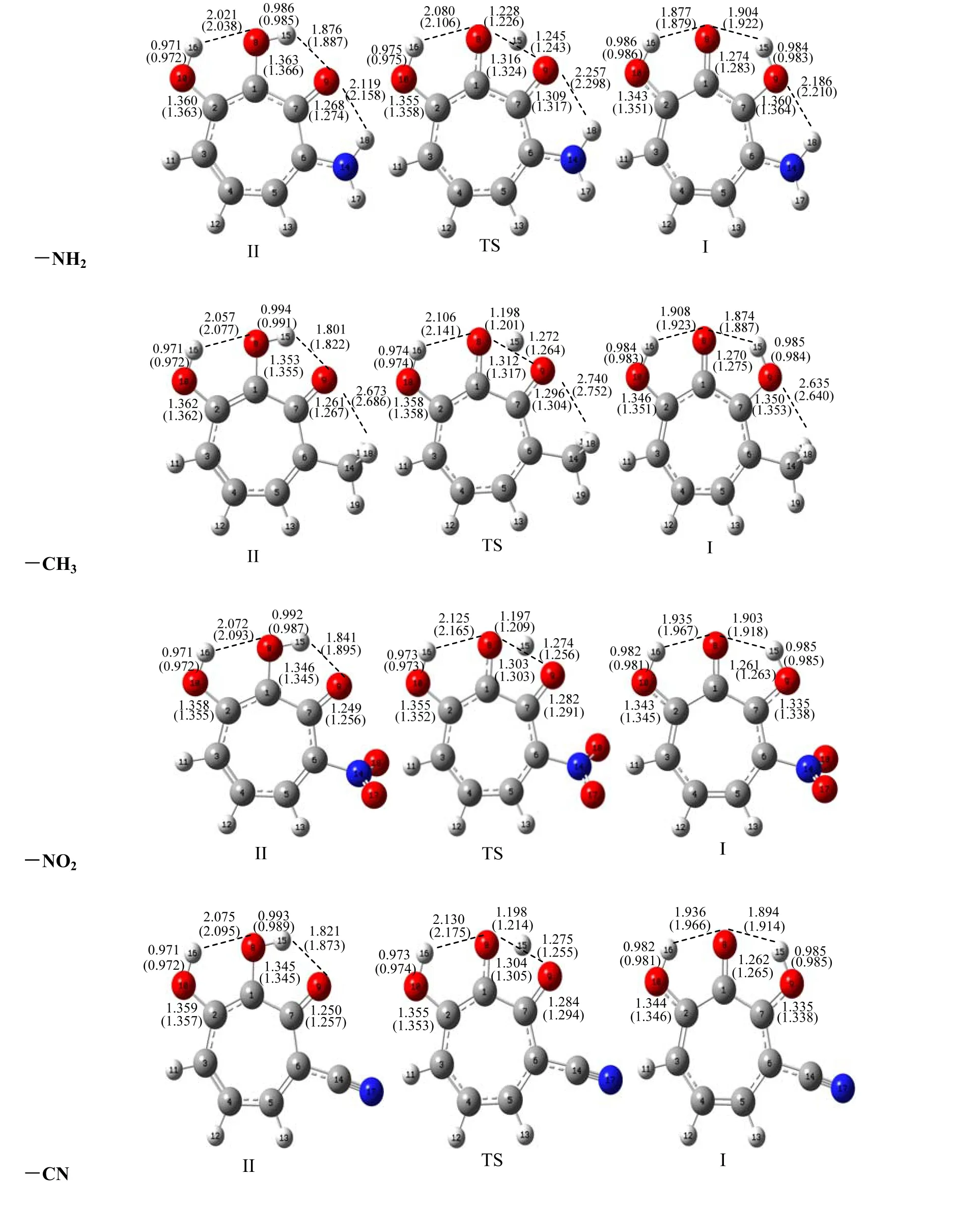
Fig. 4. Optimized structures of I, II and TS with substituents (-NH2, -CH3, -NO2 and -CN) attached to 6 position of 3-OHTRN at the B3LYP (6-31+G**) level in the gas phase. Some selected bond lengths (in Å)at the same level are given on the compounds. The numbers in parentheses belong to the aqueous phase
The proton transfer reaction was considered by II→TS→I. The transfer of hydrogen atom from the O8to the O9atom is companied by a rearrangement of the seven-membered ring, and substantial changes are observed in the carbon-carbon, carbon-oxygen and oxygen-hydrogen bonds. As seen in Tables 1 and 2, in the proton transfer II→TS→I, the C1–C7and C1–C2distances in the investigated 3-OHTRN derivatives decrease in the gas phase and in water.The C1–C7bond lengths in structures with -NH2and-CH3groups substituted on positions 3 and 5 are longer than in those substituted on positions 4 and 6 in the gas phase and in water; whereas, the C1–C2bond lengths in structures with the same groups substituted on positions 3 and 5 are shorter than in those substituted on positions 4 and 6 both in the gas phase and in water. In addition, the C1–C7and C1–C2bond lengths in structures with -NO2and -CN groups do not change with the position of substituent. One of the most striking structural features of the investigated structures is the change of C7–O9and C1–O8bond lengths. In the proton transfer II→TS→I,the C1–O8bond lengths are shortened while C7–O9are lengthened in both gas phase and water. The C1–O8bond lengths in structures (I, II and TS) with-NH2and -CH3groups are longer than those with-NO2and -CN groups. The C1–O8–H15angles in all derivatives decrease on II→I transfer, while the O8–C1–C7angles increase in the gas phase and in water. The calculated tautomerization energies (ΔE),activation energy barriers (ΔE#), free energies (ΔG),and activation free barriers (ΔG#) for the proton transfer reaction of investigated derivatives are compared in Table 3. Let’s see how the calculated relative free energies (ΔG) for the proton transfer reactions change by the position of the substituents attached to the 3-OHTRN ring. As can be seen in the results, in gas phase and water, the ΔG order of structures with -NH2and -CH3groups is 6 > 5 > 3 > 4 in terms of position of the substitution. In addition, the ΔG order of structures with -NO2and -CN groups is 6> 4 > 3 > 5 according to the position of substitution.That is, the proton transfer reactions are easier by substitution on position 5 of the electron withdrawing groups (-NO2and -CN) than on other positions of the same groups both in the gas phase and in water.
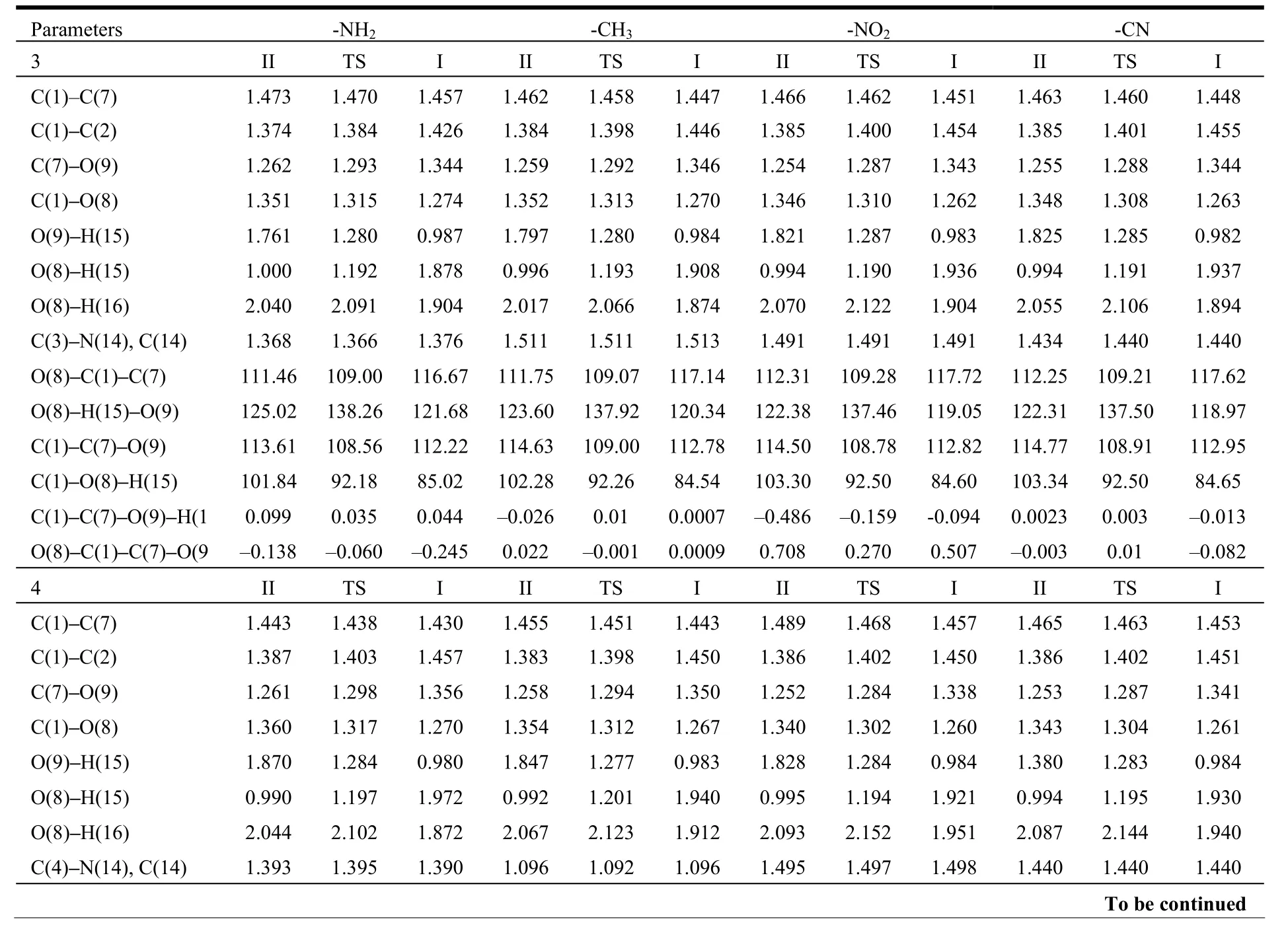
Table 1. Selected Bond Lengths (Å), Bond Angles (°) and Dihedral Angles (°) for I, II and TS Structures with Substituent Attached to 3 and 4 Positions of 3-OHTRN at the B3LYP (6-31+G**)Level in the Gas Phase (For Numbering of Atoms, See Scheme 1)
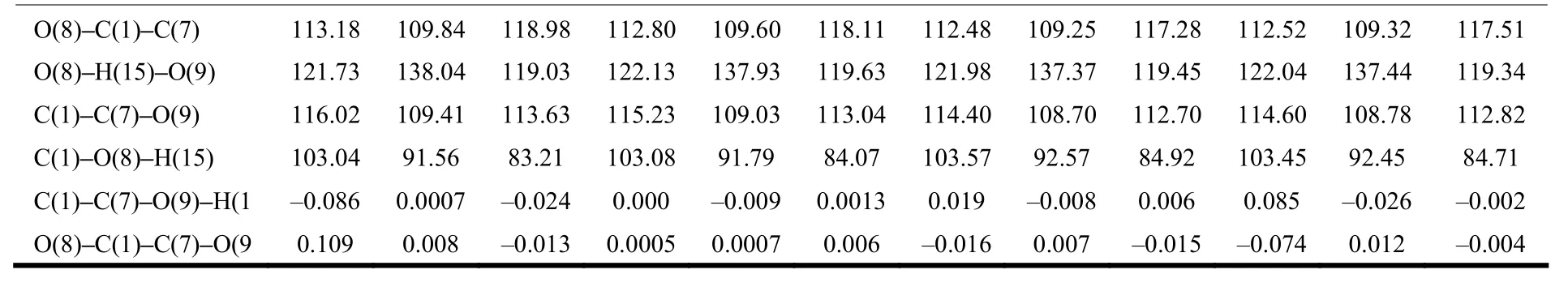
O(8)–C(1)–C(7) 113.18 109.84 118.98 112.80 109.60 118.11 112.48 109.25 117.28 112.52 109.32 117.51 O(8)–H(15)–O(9) 121.73 138.04 119.03 122.13 137.93 119.63 121.98 137.37 119.45 122.04 137.44 119.34 C(1)–C(7)–O(9) 116.02 109.41 113.63 115.23 109.03 113.04 114.40 108.70 112.70 114.60 108.78 112.82 C(1)–O(8)–H(15) 103.04 91.56 83.21 103.08 91.79 84.07 103.57 92.57 84.92 103.45 92.45 84.71 C(1)–C(7)–O(9)–H(1 –0.086 0.0007 –0.024 0.000 –0.009 0.0013 0.019 –0.008 0.006 0.085 –0.026 –0.002 O(8)–C(1)–C(7)–O(9 0.109 0.008 –0.013 0.0005 0.0007 0.006 –0.016 0.007 –0.015 –0.074 0.012 –0.004

Table 2. Selected Bond Lengths (Å), Bond Angles (°) and Dihedral Angles (°) for I, II and TS Structureswith Substituent Attached to 5 and 6 Positions of 3-OHTRN at the B3LYP(6-31+G**) Level in the Gas Phase (For Numbering of Atoms, See Scheme 1)
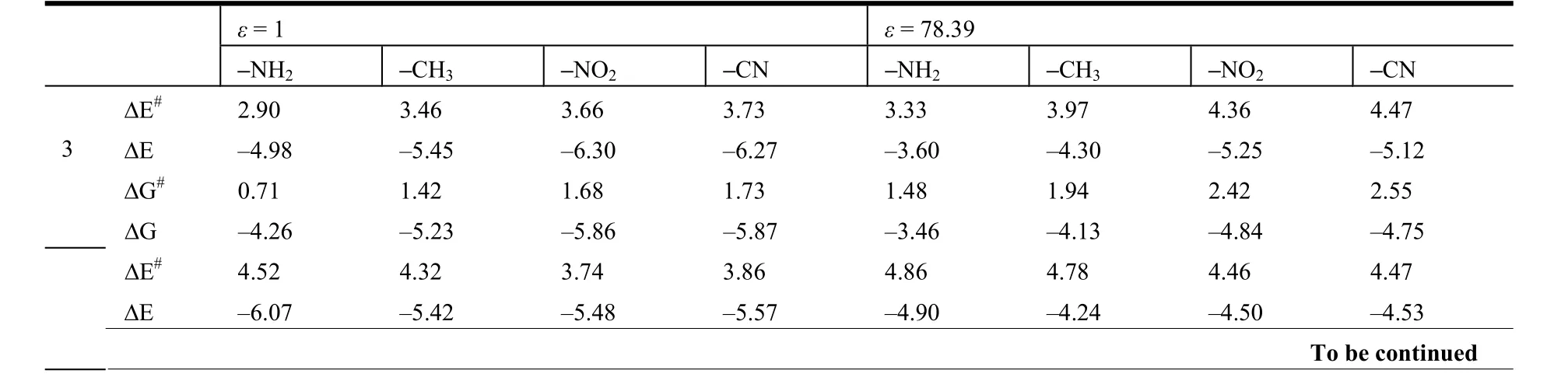
Table 3. Calculated Tautomerization Energies (ΔE), Activation Energy Barriers (ΔE#), Free Energies (ΔG) and Activation Free Energy Barriers (ΔG#) (in kcal/mol) for the Proton Transfer Reactions of 3-OHTRN Derivatives in the Gas Phase and Water

ΔG# 2.49 1.96 1.77 1.88 2.92 2.25 2.55 2.53 ΔG –5.73 –5.33 –5.09 –5.17 –4.66 –4.32 –4.12 –4.20 ΔE# 3.51 3.97 3.64 3.70 3.67 4.42 4.21 4.28 ΔE –4.05 –5.12 –7.22 –6.54 –3.05 –3.98 –6.31 –5.56 ΔG# 1.45 1.56 1.87 1.73 1.02 1.76 3.55 2.35 5 ΔG –3.70 –4.98 –6.32 –6.12 –2.55 –4.06 –5.92 –5.19 ΔE# 6.26 3.85 4.16 4.01 5.97 4.08 5.10 5.11 ΔE –1.32 –4.47 –4.32 –4.59 –1.46 –3.80 –2.29 –2.58 ΔG# 4.29 1.71 2.34 2.03 3.25 1.96 3.46 3.15 6 ΔG –0.74 –4.45 –3.87 –4.17 –1.44 –3.47 –1.84 –2.25
However, by substitution on position 4 of electron donating groups (-NH2and -CH3), the reactions are easier than by substitution on other positions of these groups. In addition, the proton transfer reactions become difficult by substitution on position 6 of each of these groups. The calculated ΔG and ΔG#values in Table 3 show that these reactions are either thermodynamically or kinetically easier in the gas phase than in water, except the reaction of structure with -NH2group at position 6. The reason of this exception is magnitude of dipole moments of I, II and TS structures with -NH2group at position 6 in water.The structures with large dipole moments could be expended to be more stable than those having smaller dipole moments in an environment with high dielectric constant. The dipole moments of I and TS structures with -NH2group at position 6 are higher than the II structure with the same position and group.As also seen in ΔEs(solvation energies, Table 4), II and TS structures with -NH2group at position 6 become more stable than the I structure with the same group and position in water due to the solvent effects.Therefore, it can explain that the energy barrier of proton transfer reaction for structure with -NH2group at position 6 decreases and that the reaction is also thermodynamically easier in water than in gas phase.
Let’s see also how the calculated free energies (ΔG)for the proton transfer reactions change by different substituents attached on the same position. While the reaction is the easiest by substitution of the -CN group(ΔG value for the reaction with -NH2group at the same position is only higher than those with -CN group by 0.01 kcal/mol in the gas phase) among the groups attached to position 3, the proton transfer for structure with -NH2group at position 4 is the easiest among the groups attached to position 4 in the gas phase and in water. In addition, the proton transfer is the easiest by substitution of -NO2group on position 5 and -CH3group on position 6 among the groups attached to the same position both in gas phase and in water.
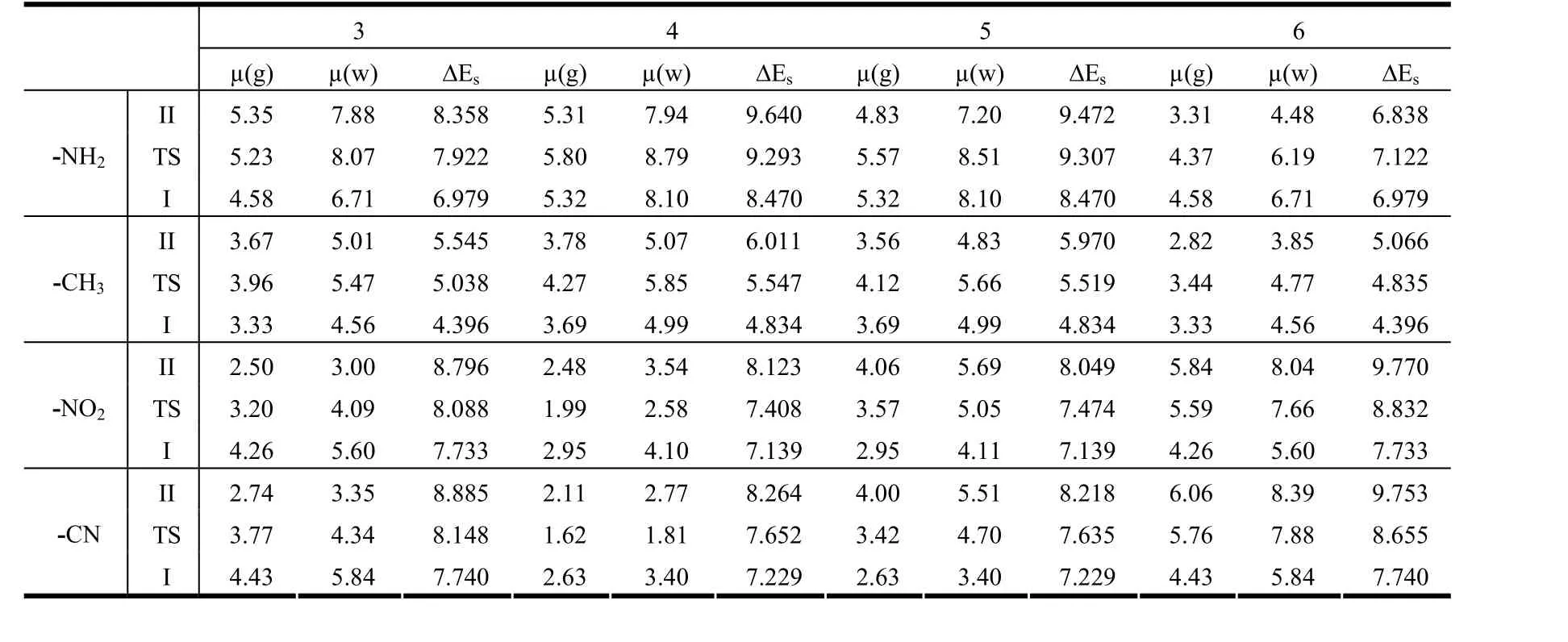
Table 4. Dipole Moments (µ(g) in Gas Phase and µ(w) in Water, in Debye) and Solvation Energies ΔEs = Egas – Esolv (kcal/mol) of the Tautomeric Forms in Gas Phase and Water
From the viewpoint of valence theory, the interaction between the lone pair of acceptor O atom and O–H σ*orbital is mainly responsible for the proton transfer. Therefore, the O···H–O angle and O···H distance may play important roles in the proton transfer reactions. In most cases, hydrogen bonds with linear O···H–O structures and short hydrogen bond distances are considered to be strong. In addition, the O8···H15distances in TS structures have important effect on the energy barriers of the reactions.According to this information, evaluated to activation free energy barrier (ΔG#) for -NH2substituted systems,the ΔG#order is 6 > 4 > 5 > 3 in terms of substitution position in the gas phase. The same order is 6 > 4 > 3> 5 in water. Hence, in the case attached to the -NH2group at position 3, the ΔG#value for the reaction is the lowest in the gas phase. Comparing the above mentioned bond lengths and bond angles in the II and TS structures, it can be seen that the II structure with-NH2group at position 3 has the most linear O8···H15-O9structure and the shortest O8···H15hydrogen bond. Also, the O8···H15distance is the shortest in the TS structure with -NH2group in the same position (Fig. 1). In water, the ΔG#is lower for structure with -NH2group at position 5 than that with the same group at position 3. Because the dipole moment of the TS structure with -NH2group at position 5 is larger than that with the -NH2substituted on position 3, the TS structure with -NH2group at position 5 becomes stable in water, while the ΔG#order for -CH3substituted systems in the gas phase is 4 > 6 > 5 > 3 in terms of substitution position, and 4 >6 > 3 > 5 in water. That is, both in gas phase and water,the reaction having -CH3group at position 4 is the most difficult. As seen in Table 2, the II structure with-CH3group at position 4 has the weakest O9···H15hydrogen bond. Also, the TS structure with -CH3group at position 4 has the longest O8···H15distance among further TS structures with -CH3group (see Fig. 2).The ΔG#order for -NO2substituted systems is 6 > 5 >4 > 3 in terms of substitution position in the gas phase.In water, the same order is 5 > 6 > 4 > 3. That is, in both gas phase and water, the ΔG#is the lowest for system with -NO2group at position 3. The II and TS structures with -NO2group at position 3 have the shortest hydrogen bonds. The ΔG#order for -CN substituted systems is 6 > 4 > 5 = 3 in terms of substitution position in the gas phase. In water, the same order is 6 > 3 > 4 > 5. The proton transfer in structures with -CN group at position 5 is the easiest in gas phase and water.
4 CONCLUSION
The major conclusions to be gleaned from this work are as follows,
(1) Tautomer I structures are more stable than tautomer II structures in gas phase and in water.
(2) The calculated ΔG and ΔG#values show that these reactions are either thermodynamically or kinetically easier in gas phase than in water, except the reaction of structure with -NH2group at position 6.
(3) Comparing the calculated free energies (ΔG)for the proton transfer reactions of structures with the same substituent attached to different positions, the proton transfer reactions become difficult by substitution on position 6 of each group in gas phase and in water.
(4) The proton transfer reactions are easier by substitution on position 5 of electron withdrawing groups (-NO2and -CN) than substitution on other positions of the same groups in both gas phase and water. On the other hand, by substitution on position 4 of electron donating groups (-NH2and -CH3), the reactions are easier than substitution on other positions of these groups.
(5) Considering ΔG and ΔG#values for all investigated reactions, the proton transfer reaction is kinetically the easiest by substitution on position 3 of-NH2group in the gas phase but, it is kinetically the easiest by substitution on position 5 of the same group in water. Also, the proton transfer reaction is thermodynamically the easiest by substitution of-NO2group on position 5, but it is thermodynamically the most difficult by substitution on position 6 of -NH2group in the gas phase and in water.
ACKNOWLEDGEMENTS The authors acknowledge the CUBAP (The Scientific Research Projects Council of Cumhuriyet University) for providing the Gaussian 03W and Gauss View 3.0 program packages.
(1) Camilo, A.; Alves, P.; Hollas, J. M.; Hamdan, M.; Ridley, T. The 370-nm electronic spectrum of tropolone: evidence from single vibronic level fluorescence spectra regarding the assignment of some vibrational fundamentals in the X~ and à states. J. Mol. Spectrosc. 1985, 109, 99-122.
(2) Sekiya, H.; Nagashima, Y.; Nishimura, Y. Vibrational specificity of proton-transfer dynamics in ground-state tropolone. Bull. Chem. Soc. Jpn. 1989, 62,3229-3231.
(3) Yi, P. G.; Liang, Y. H.; Tang, Z. Q. Theoretical study of intermolecular proton transfer reaction in isolated 5-hydroxyisoxazole-water complexes. Chem.Phys. 2006, 322, 387-391.
(4) Casadesus, R.; Moreno, M. A theoretical study of the ground and first excited singlet state proton transfer reaction in isolated 7-azaindole-water complexes. Chem. Phys. 2003, 290, 319-336.
(5) Ikegami, Y. The infrared spectra of troponoid compounds. VI. The infrared and Raman spectra of tropolone, 3- and 4-isopropyltropolones. Bull. Chem.Soc. Jpn. 1963, 36, 1118-1125.
(6) Alves, A. C. P.; Hollas, J. M. The near ultra-violet absorption spectrum of tropolone vapour II. Vibrational analysis. Mol. Phys. 1973, 25, 1305-1314.(7) Redington, R. L.; Redington, T. E. Tropolone monomer: vibrational spectrum and proton tunneling. J. Mol. Spectrosc. 1979, 78, 229-247.
(8) Tomioka, Y.; Ito, M.; Mikami, N. Electronic spectra of tropolone in a supersonic free jet. Proton tunneling in the S1 state. J. Phys. Chem. 1983, 87,4401-4405.
(9) Redington, R. L.; Chen, Y.; Scherer, G. J.; Field, R. W. Laser fluorescence excitation spectrum of jet-cooled tropolone. J. Chem. Phys. 1988, 88,627-633.
(10) Sekiya, H.; Nagashima, Y.; Nishimura, Y. Electronic spectra of jet-cooled tropolone. Effect of the vibrational excitation on the proton tunneling dynamics. J. Chem. Phys. 1990, 92, 5761-5769.
(11) Sekiya, H.; Nagashima, Y.; Tsuji, T.; Nishimura, Y.; Mori, A.; Takeshita, H. Vibrational mode-specific tunneling splittings in the A states of deuterated tropolones. J. Phys. Chem. 1991, 95, 10311-10317.
(12) Nishi, K.; Sekiya, H.; Kawakami, H.; Mori, A.; Nishimura, Y. Coupling of internal rotation of methyl group with proton transfer in the S1 state of 5-methyltropolone. J. Chem. Phys. 1998, 109, 1589-1592.
(13) Frost, K.; Hagemeister, F. C.; Arrington, C. A.; Zwier, T. S.; Jordan, K. D. Fluorescence-dip infrared spectroscopy of tropolone and tropolone-OD. J.Chem. Phys. 1996, 105, 2595-2604.
(14) Mó, O.; Yáñez, M.; Esseffar, M.; Herreros, M.; Notario, R.; Abboud, J. L. Role of chelation and resonance on the intrinsic acidity and basicity of tropolone. J. Org. Chem. 1997, 62, 3200-3207.
(15) Redington, R. L.; Bock, C. W. MO study of singlets, triplets, and tunneling in tropolone. 1. Geometries, tunneling, and vibrations in the ground electronic state. J. Phys. Chem. 1991, 95, 10284-10294.
(16) Sanna, N.; Ramondo, F.; Bencivenni, L. MP2/6-31G* structural study of tropone and tropolone molecules. J. Mol. Struct. 1994, 318, 217-235.
(17) Nash, J. J.; Zwier, T. S.; Jordan, K. D. Mode-selective photoisomerization in 5-hydroxytropolone. II. Theory. J. Chem. Phys. 1995, 102, 5260-5270.
(18) Vener, M. V.; Scheiner, S.; Sokolov, N. D. Theoretical study of hydrogen bonding and proton transfer in the ground and lowest excited singlet states of tropolone. J. Chem. Phys. 1994, 101, 9755-9765.
(19) Smerdachina, Z.; Siebrand, W.; Zgierski, M. Z. Mode-specific hydrogen tunneling in tropolone: an instanton approach. J. Chem. Phys. 1996, 104,1203-1212.
(20) Takada, S.; Nakamura, H. Effects of vibrational excitation on multidimensional tunneling: general study and proton tunneling in tropolone. J. Chem.Phys. 1995, 102, 3977-3992.
(21) Wójcik, M. J.; Boczar, M.; Stoma, M. Spectroscopic and theoretical study of vibrational spectra of hydrogen-bonded tropolone. Int. J. Quantum.Chem. 1999, 73, 275-282.
(22) Tsuji, T.; Hamabe, H.; Hayashi, Y.; Sekiya, H.; Mori, A.; Nishimura, Y. Investigation of intramolecular hydrogen bonds in ortho-hydroxytropolone. J.Chem. Phys. 1999, 110, 966-971.
(23) Kubo, K.; Matsimoto, T.; Mori, A. 3-Hydroxytropolone (2,7-dihydroxycyclohepta-2,4,6-trien-1-one). Acta Cryst. 2007, E63, o941-o943.
(24) Isin, D. Ö.; Karakus, N. Computational study of the intramolecular proton transfer reactions of 3-hydroxytropolone(2,7-dihydroxycyclohepta-2,4,6-trien-1-one) and its dimers. J. Mol. Model. 2010, 16, 1877-1882.
(25) Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti conelation energy formula into a functional of the electron density. Phys. Rev. B 1988,37, 785-789.
(26) Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648-5652.
(27) Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098-3100.
(28) Foresman, J. B.; Keith, T. A.; Wiberg, K. B.; Snoonian, J.; Frich, M. J. Solvent effects. 5. Influence of cavity shape, truncation of electrostatics, and electron correlation on ab Initio reaction field calculations. J. Phys. Chem. 1996, 100, 16098-16104.
(29) Frisch, M. J. et al. Gaussian, Inc., Pittsburgh PA 2003.
(30) Frisch, A.; Dennington II, R.; Keith, T.; Nielsen, A. B.; Holder, A. J. GaussView Reference, Version 3. 0. Gaussian İnc 2003.
