
常温合成聚羧酸减水剂工艺的理论研究
2014-03-15 09:05:05王浩逄建军叶冉冉张力冉王栋民
商品混凝土 2014年5期
王浩,逄建军,叶冉冉,张力冉,王栋民
(中国矿业大学(北京),北京 100083)
综述评论
常温合成聚羧酸减水剂工艺的理论研究
王浩,逄建军,叶冉冉,张力冉,王栋民
(中国矿业大学(北京),北京 100083)
常温合成聚羧酸减水剂不仅可以有效降低生产能耗和成本,而且还能简化生产操作流程。本文介绍了自由基聚合机理,通过聚合机理推导其反应动力学,并结合反应动力学重点探讨了引发作用和引发剂。最终确定,在常温下合成聚羧酸减水剂需要采用氧化还原引发体系,增加引发剂和单体浓度,延长反应时间等措施。
聚羧酸减水剂;常温;自由基聚合;反应动力学
0 引言
聚羧酸减水剂作为第三代混凝土减水剂,与前几代混凝土减水剂相比,具有减水率高、坍落度保持好、生产过程无污染等优点[1,2]。通常,聚羧酸减水剂的合成温度为50~70℃。对于大釜生产的聚羧酸减水剂产品,反应温度直接影响了产品的质量,所以在生产过程中需着重调整釜内温度。但这样做不仅增加了工作人员的操作难度,而且与常温相比,较高的温度还增加了能耗,提高了生产成本,使得产品的竞争力降低。因此,低温(常温)合成聚羧酸减水剂的工艺被提上日程。然而,由于反应温度的降低,使得反应速率、聚合度以及分子结构等发生了改变,对聚羧酸减水剂的性能影响较大。目前常温合成工艺尚处于研究阶段,有着十分广阔的研究和应用前景。
本文介绍了自由基聚合反应机理,通过反应机理推导其反应动力学,并结合反应动力学重点介绍引发作用和引发剂。以上述理论为基础来确定常温合成聚羧酸减水剂的思路。
1 自由基聚合反应机理
1.1 链引发
第一步反应是引发剂Ⅰ均裂,产生一对初级自由基 R·。反应见式 (1),第二步是初级自由基和单体 M 加成,形成单体自由基 RM·,见式 (2)。
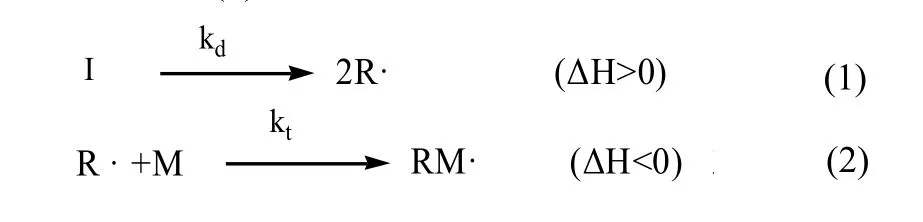
其中,引发剂分解是吸热反应,所吸收的热量等于均裂时所需的键能。反应的活化能较高,约 125kJ/mol,反应速率小。若使用高活性引发剂或氧化还原引发体系,能使反应的活化能降低,分解速率相应提高。由于它是聚合反应中速度最慢的一步,所以为控制步骤。
第二步初级自由基和单体加成反应是打开π键、重新杂化、生成σ键的过程,是放热反应,活化能较低,约为21~34kJ/mol,反应的速率比第一步快。
1.2 链增长
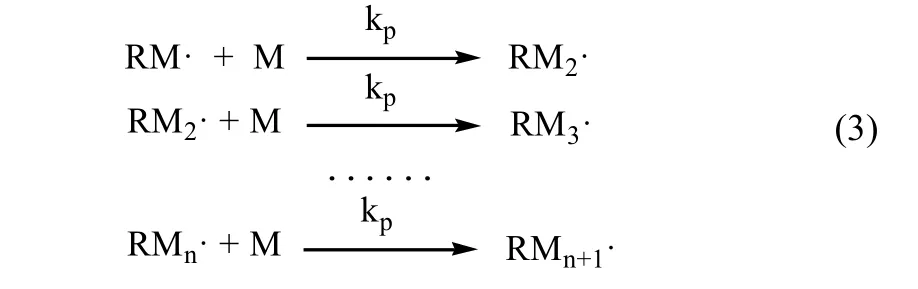
链增长是放热反应,它所需的活化能较低,约为21~34kJ/mol。具体反应见式 (3)。
1.3 链终止
链终止包括偶合终止和歧化终止。其中两大分子链自由基末端的孤电子相互成共价键,形成饱和大分子的反应称做偶合终止,见式 (4) 所示。
一个链自由基夺取另一个链自由基上的β氢原子而终止,称为歧化终止。最终将形成两个聚合物分子。夺得氢原子的大分子端基饱和,失去氢原子的则不饱和,见式 (5) 所示。

1.4 链转移
在自由基聚合反应过程中,链自由基可能从单体、引发剂、溶剂或大分子上夺取一个原子而终止,这些失去原子的分子则变为自由基,继续增长。这种把活性种转移给另一分子使反应继续下去,而原来活性种本身却终止的反应称为链转移反应。链转移主要分为:向单体转移、向引发剂转移、向溶剂转移以及向大分子转移。其中,向单体转移不会影响自由基活性,但会影响到聚合物的聚合度。而向引发剂转移会降低引发速率,不仅使聚合度降低,还会使引发剂效率下降。当向大分子转移时,会使得大分子主链上连有支链。
1.5 自由基聚合微观动力学
通过研究自由基聚合微观动力学可以掌握自由基反应进程,还可以确定反应单体、链转移剂及引发剂用量,对合成反应起指导作用。将自由基聚合微观动力学大致可分为引发剂分解动力学和聚合反应动力学。其中,通过研究引发剂分解动力学可以确定该引发剂的半衰期,通过定量的求得半衰期可确定反应时间。具体求法见式 (6)、式 (7)。
其中:
I——引发剂在 t 时刻的浓度,mol/L;
I0——引发剂初始浓度,mol/L;
T——时间,s;
kd——引发分解速率常数,其与温度有关,即kd= Ad e-Ea/RT。测定不同温度下的 kd,通过作图即可求得指前因子 Ad以及反应活化能 Ea。
在研究和推导聚合反应动力学前,通常有三个假定[3]:
(1)高分子的聚合度很大,即引发时消耗的单体所占的比例很小,可以忽略;
(2)等活性理论,即增长反应中各步速率常数相同;
(3)“稳态”,即产生自由基的引发速率与自由基消失的终止速率相等,或体系中自由基浓度恒定。
引发剂的分解速率最慢,是控制步骤,如前所述,1mol的引发剂可分解为 2mol 初级自由基,由于诱导分解和笼蔽效应使引发过程伴随副反应,所以必须考虑引发剂的引发效率,故链引发速率见式 (8)。通过等活性理论可得;链自由基的活性与链增长基本无关,所以其速率方程见式 (9)。链终止方式分为偶合终止和歧化终止,通常情况下,聚羧酸减水剂聚合反应属于偶合终止,链终止反应速率方程见式 (10)。由于经过一段聚合时间后,引发速率 (Ri) 与终止速率相等(Rtc),联系两式,可得[M.]=(2Rki)1/2。同时由于高分子聚合度很大,用于引发的单体远小于链增长所消耗的单体,故聚合总速率等于链增长速率。所以结合式 (8)、式 (9) 和式 (10) 可聚合总反应速率方程,见式 (11)。
其中:

kp——链增长速率常数;
F——引发效率;
Ad——指前因子;
Ea——反应活化能,kJ/mol;
kt——链终止速率常数;
[M]、[I]、[M·]——分别为单体、引发剂和单体自由基浓度,mol/L。
通过反应速率通式可以得出:聚合总反应速率与单体浓度、引发剂浓度以及反应温度等因素有关。对于聚羧酸减水剂合成体系,在其他因素不变的条件下,随着引发剂及单体浓度的增加,总聚合反应速率增加;若在常温下聚合,由于温度的降低,使得聚合反应速率减小。所以在常温下合成可通过增加引发剂和反应单体浓度、降低聚合反应活化能来增加反应速率,也可采用延长反应时间的方法来提高聚合反应的单体转化率。但是,如果反应初期引发剂的浓度过高,会导致副产物的增多,甚至使单体产生交联作用,所以引发剂浓度还需控制在一个合适的范围内。
2 水溶性引发剂种类
聚羧酸减水剂的合成通常属于水溶性聚合反应,故在下文主要介绍水溶性引发和水溶性引发剂的相关信息。
2.1 无机过氧化物
2.1.1 过氧化氢
过氧化氢是最简单的过氧化物,其中一个或两个氢原子被取代,可以衍生出许多过氧化氢物和过氧化物。过氧化氢均裂的结果,形成两个氢氧自由基。
HO-OH→2HO·
其分解的活化能较高,约为 220kJ/mol,须在较高温度下方能分解。所以一般不单独用作为引发剂,主要用于氧化还原引发体系中。
2.1.2 过硫酸盐
过硫酸盐也是常用的无机过氧化物,例如过硫酸钾(K2S2O8) 和过硫酸铵 (NH4)2S2O8。他们属于水溶性引发剂,其中过硫酸钾的结构式如图 1 所示[4]:
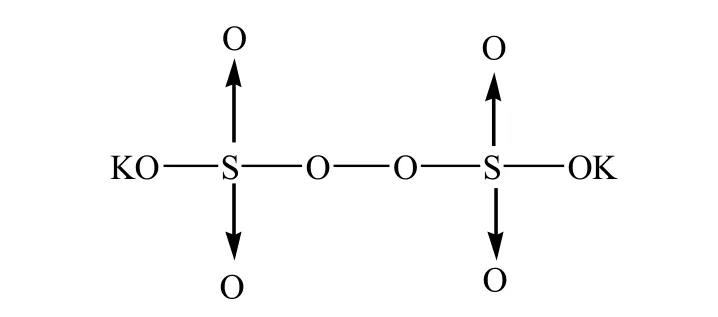
图 1 过硫酸钾的化学结构式
过硫酸盐在水溶液中按一级反应分解成过硫酸根自由基,然后进一步引发单体。
S2O82-→2HSO4-·
在碱性、中性和弱酸性溶液中,过硫酸盐分解均呈一级反应。其中在酸性溶液中分解的速率常数随离子强度增加而降低。在离子强度恒定不变条件下,pH 对 K2S2O8一级分解速率常数的影响如图 2 所示[5]。
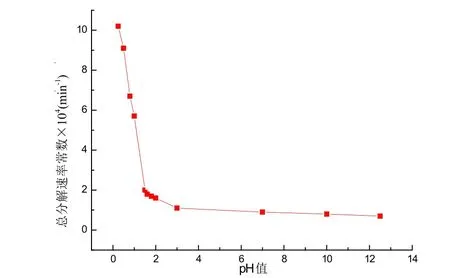
图 2 pH 值对 K2S2O8分解速率常数的影响,50℃
从图 2 可得:当 pH>3 时,pH 对分解速率常数 kd影响不大;当 pH<3 时,kd则随 pH 的降低而迅速增高。所以在过硫酸铵引发体系中,随着酸的加入,可以有效提高引发剂的分解速率。
2.2 氧化还原引发体系
过氧类引发剂加入少量还原剂,组成氧化—还原体系,通过电子转移反应,生成自由基中间产物而引发聚合。氧化还原引发体系的活化能较低,可使引发剂分解速率和聚合速率大大提高,并使诱导期缩短,在较短的时间内,就可以得到较高的转化率和较高的分子量。聚合可在室温或更低的温度下进行。表 1 所示为氧化还原引发体系反应活化能。

表 1 氧化还原引发体系反应活化能
通过表 1 中可以看出:一元引发体系中活化能是氧化还原引发体系的 3~4 倍,构成氧化还原引发体系可使活化能大大降低。所以在低温(常温)聚合时,需采用氧化还原体系引发。
对于水溶性引发体系的氧化剂有过氧化氢、过硫酸盐等,还原剂则有亚铁盐、亚硫酸钠、连二亚硫酸钠、亚硫酸氢钠、硫代硫酸钠等。氧化剂和还原剂的不同组合,就形成多种多样的氧化还原引发体系。
2.3 过硫酸盐—脂肪胺体系
这类引发体系主要用于含水介质中的聚合反应。研究发现能与过硫酸盐匹配的脂肪胺,可以是开链的伯、仲、叔三种胺,也可以是环状的脂肪仲、叔胺,或各种多元胺[6-9]。其中活性最高的是 N,N,N',N'—四甲基乙二胺 (TMEDA)。过硫酸盐—TMEDA 体系能在室温引发丙烯酰胺等聚合,用于制备聚丙烯酰胺水凝胶或聚 (N—异丙基丙烯酰胺) 温度敏感性水凝胶。
3 引发剂的选择和用量
引发剂的选择,首先根据聚合实施方法,从溶解度角度确定引发剂类型。本体聚合、悬浮聚合、有机溶液聚合选用偶氮类和过氧类等油溶性有机引发剂。乳液聚合和水溶液聚合则选用过硫酸盐类的水溶性引发剂。其次,根据聚合温度选择半衰期适当的引发剂,使聚合时间适中。
如果引发剂活性过低,则分解速度过低,需要延长聚合时间或提高反应温度。相反,若引发剂活性过高,半衰期过短,虽然可以提高反应速率,但短时间会放出大量聚合热,温度往往不易控制,甚至引发爆聚。另一方面,引发剂还可能过早分解完毕,在低转化率阶段就停止聚合。根据经验,可以根据氯乙烯聚合时间约等于半衰期三倍的规则,来选用引发剂[10]。引发剂一般只引发聚合,与生成聚合物的结构无关。引发剂残基接在大分子链末端,所占的比例虽小,但在考虑毒性、大分子端基的反应活性等情况时应加以注意。
4 小结
根据聚合反应速率通式可得:聚合反应速率与链引发速率常数、链增长速率常数、引发效率以及单体和引发剂的浓度成正比,与链终止速率常数成反比。由于反应单体和引发剂浓度便于调整,故常温合成减水剂可考虑单体及引发剂浓度对合成产物的影响;通过理论分析可得降低聚合反应活化能可有效增加聚合反应速率,所以还可尝试调整聚合体系pH、寻求引发活性高的引发剂以及采用氧化还引发原体系等方法来降低反应活化能。通过上述方法可使常温聚合时反应产物的聚合速率接近高温聚合时的速率,结合上述结论可对常温合成聚羧酸减水剂提供理论基础。
[1] 孙振平,黄雄荣.烯丙基聚乙二醇系聚羧酸减水剂的研究[J].建筑材料学报,2009,12(4): 407-412.
[2] 翁荔丹,黄雪红.聚羧酸减水剂对水泥水化过程的影响[J].福建师范大学学报(自然科学版),2007,23(1): 54-57.
[3] 潘祖仁.高分子化学[M].北京:化学工业出版社,2010:82-84.
[4] 王子明.混凝土高效减水剂[M].北京:化学工业出版社, 2011:257-262.
[5] I.M.Kolthoff, I.K.Miller.The Chemistry of Persulfate. I. The Kinetics and Mechanism of the Decomposition of the Persulfate Ion in Aqueous Medium[J]. Journal of the American Chemical Society, 1951, 73 (7):3055-3059.
[6] X.D Feng,X.Q.Guo,K.Y.Qiu.Studies on the initiation mechanism of persulfate/aliphatic secondary amine system in vinyl polymerization[J].Polym Bull,1987, 18(1):19-26.
[7] Feng X D,Guo X Q,Qiu K Y.Study of the initiation mechanism of the vinyl polymerization with the system persulfate/N,N,N',N'-tetramethylethylenediamine[J]. Makromol Chem,1988,189(1):77-83.
[8] Guo X Q,Qiu K Y. feng X D.Studies on the kinetics and initiation mechanism of acrylamide polymerization using persulfate/aliphatic diamine systems as initiator[J].Makromol Chem,1990, 191(3): 577-587.
[9] 郭新秋,丘坤元,冯新德.过硫酸盐和脂肪二元叔胺体系引发烯类聚合的反应与机理的研究[J].中国科学(B 辑),1989, (1-12): 1134-1142.
[10] 潘祖仁,于在璋.自由基聚合[M].北京:化学工业出版社,1983: 100-101.
[通讯地址]北京市海淀区中国矿业大学(北京)化学与环境工程学院(100083)
王浩(1990—),男,在读硕士研究生,主要从事聚羧酸减水剂结构设计与优化方面的研究。
猜你喜欢
公路与汽运(2024年1期)2024-03-07 03:02:06
河南科技(2023年1期)2023-02-11 12:17:04
钻井液与完井液(2022年4期)2022-10-26 06:39:38
Chinese Physics B(2022年5期)2022-05-16 07:10:38
云南化工(2021年10期)2021-12-21 07:33:28
哈尔滨轴承(2021年1期)2021-07-21 05:43:14
建材发展导向(2021年24期)2021-02-12 02:00:02
黑龙江交通科技(2020年5期)2020-01-13 18:01:35
电镀与环保(2017年3期)2017-06-23 08:24:51
中国塑料(2014年1期)2014-10-17 02:46:34
