
神经营养因子-3对周围神经损伤后肌细胞凋亡及Ca2+-ATP酶的影响*
2014-03-15 05:49宗海斌黄媛霞董玉珍周慧聪
重庆医学 2014年3期
宗海斌,黄媛霞,董玉珍△,周慧聪
(1.新乡医学院基础医学院机能实验室,河南新乡 453003;2.新乡医学院第一附属医院骨外科,河南卫辉 453100)
周围神经损伤后肌细胞凋亡导致不可逆性失神经肌萎缩,在神经恢复前有效地抑制细胞的凋亡减缓肌萎缩,一直是中枢、周围神经损伤治疗的一个重要方面[1]。许多学者认为周围神经损伤后神经营养因子分泌减少,给予外源性神经营养因子-3(neurotrophin 3,NT-3)对促进神经再生及保护肌萎缩均有作用[2],但其作用机制却不清楚。本实验应用NT-3治疗大鼠坐骨神经损伤模型,通过观察治疗后其对caspase-3基因、肌细胞凋亡、Ca2+-ATP酶等指标的影响,来探讨NT-3 对神经损伤修复和肌萎缩的保护作用机制。
1 材料与方法
1.1材料 Wistar 成年大鼠60只由新乡医学院动物实验中心提供,雌雄不拘,体质量200~250 g;S-100免疫组织化学染色、细胞蛋白提取、凋亡细胞试剂盒均由湖北武汉博士德生物工程有限公司提供;NT-3 质粒由北京塞百盛生物工程有限公司合成提纯。恒温培育箱、离心机、冰冻切片机购自德国Leica公司;分子生物学凝胶电泳仪购自美国Sigma公司;TJTY-300图像分析软件及光镜购自日本Philips 公司。
1.2实验方法
1.2.1动物模型制作、分组 健康Wistar大鼠60只,2%戊巴比妥钠腹腔内注射麻醉(30 mg/kg),在16倍显微镜下暴露坐骨神经,距坐骨神经出口下2 cm处切断,近端返折缝合到附近肌肉当中,远断端用10-0显微缝线结扎,建立坐骨神经损伤动物模型。分组后使用微量注射器腓肠肌内分别注射生理盐水和NT-3基因2 μL(浓度为1 μg/μL),注射后留针2 min,术后所有动物按常规分笼饲养。
1.2.2测定caspase-3蛋白表达 于术后2 h、4 h、8 h、1 d、3 d、7 d、14 d和21 d麻醉下抽取腓肠肌总RNA,采用细胞蛋白提取试剂盒(将肌细胞内的caspase-3蛋白完全提取)进行PCR,反应体系(取2 μL反转录产物为模板,按常规聚合酶链反应的条件,加入Taq聚合酶0.5 μL,内参照(actin)引物3 μL,先将样品于94 ℃变性5 min,按下列参数循环35次:94 ℃变性1 min,55 ℃退火1 min,55 ℃延伸1 min;最后72 ℃反应10 min)扩增行凝胶电泳;caspase-3 引物序列包括caspase-3上游引物:5′-AAG AAG ACC ATA GCA AAA GGA G-3′,下游引物:5′-CAC AAA GTG ACT GGA TGA ACC-3′;内参照β-actin 上游引物:5′-CCA A GG CCA ACC GCG AGA AGA TGA C-3′,下游引物:5′-AGG GTA CAT GGT GGT GCC GCC AGA C-3′。腓肠肌行石蜡切片加caspase-3抗体染色、封片,光镜下观察:50 μL的10%山羊血清(25 ℃) 30 min,50 μL caspase-3兔多克隆抗体(浓度为1∶200), 4 ℃过夜,50 μL二抗(浓度为1∶150)25 ℃ 20 min,50 μL HRP标记链亲和素(25 ℃)10 min,二氨基联苯胺(DAB)显色,苏木精复染,阳性信号以胞质出现棕黄进行呈色反应,阴性对照,应用TJTY-300图像分析软件测定caspase-3蛋白表达的水平。
1.2.3腓肠肌细胞凋亡检测 于术后1、4、8周分别取各组失神经腓肠肌标本制成厚5 μm石蜡切片,荧光TUNEL法检测凋亡细胞,严格按TUNEL凋亡原位检测试剂盒说明书进行操作,被标记的阳性细胞核呈棕黄色。每张切片选取10个不重复视野计数阳性细胞数,按下列公式计算细胞凋亡率。细胞凋亡率=阳性细胞核数/总细胞核数×100%,取平均数计算。
1.2.4Western blot法检测肌质网Ca2+-ATP酶 术后1、4、8周将腓肠肌标本取等量蛋白采用凝胶电泳分离,转膜后用25 mL封闭液室温封闭1 h,加入一抗孵育3 h,再加入辣根过氧化物酶标记的二抗,压片、冲片后采用化学系统检测各组Ca2+-ATP酶表达量。

2 结 果
2.1caspase-3基因表达 caspase-3基因表达的变化扩增产物行琼脂糖凝胶电泳,对含插入NT-3基因片段的产物进行硬胶回收,并对回收产物序列进行酶切鉴定,可见各目的基因及内参条带清晰,见图1、2。

图1 生理盐水组caspase-3电泳表达

图2 NT-3 组caspase-3电泳表达
2.2caspase-3蛋白表达 两组caspase-3蛋白表达在损伤2 h后即开始升高,随时间增长继续升高;NT-3组在术后7 d增长比率逐渐下降,caspase-3蛋白的表达2 h后低于生理盐水组(P<0.05)。见表1,图3、4。

图3 21 d对照组caspase-3蛋白表达(×200)
2.3肌细胞凋亡率比较 两组术后1周均见肌细胞凋亡,比较差异无统计学意义(P>0.05);4、8周生理盐水组凋亡细胞明显增多,高倍镜下见细胞缩小,形状不规则,核膜皱缩,核内出现棕黄色颗粒,而NT-3组阳性细胞明显减少。见表2,图5、6。

表1 各时间点两组caspase-3蛋白表达比较
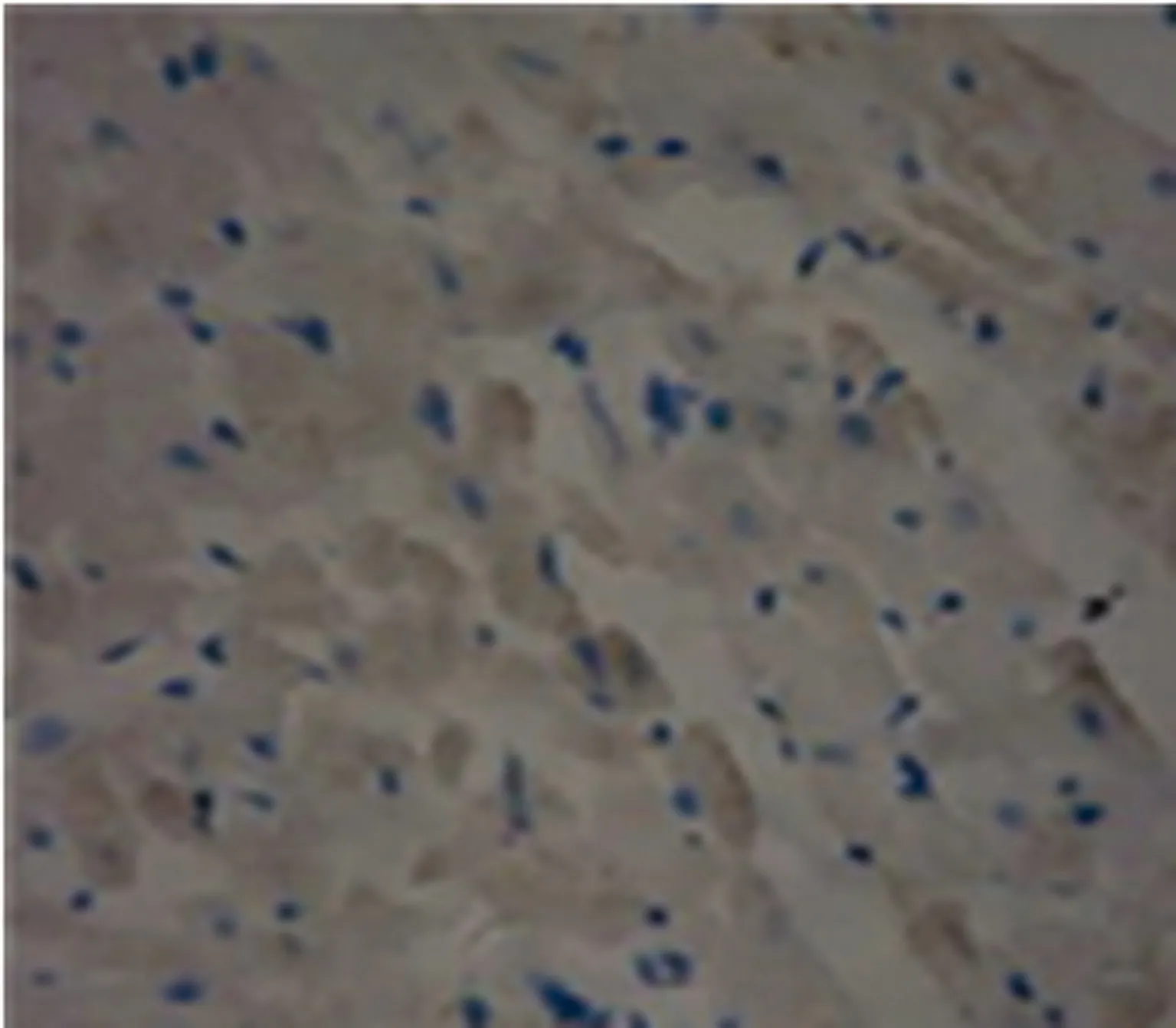
图4 21 d NT-3组caspase-3蛋白表达(×200)

图5 8周生理盐水组肌细胞凋亡情况(×400)

图6 8周NT-3组肌细胞凋亡情况(×400)
2.4两组Ca2+-ATP酶变化 1周生理盐水组Ca2+-ATP酶水平与NT-3组差别不大,而4、8周比较差异有统计学意义(P<0.05)。

图7 Western blot法Ca2+-ATP酶检测结果
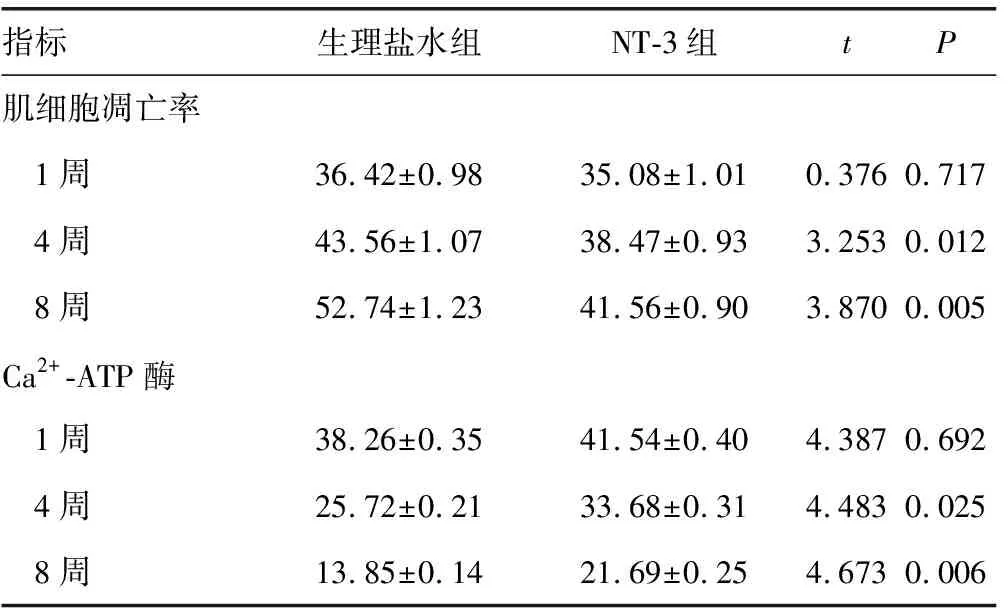
指标生理盐水组NT-3组tP肌细胞凋亡率 1周36.42±0.9835.08±1.010.3760.717 4周43.56±1.0738.47±0.933.2530.012 8周52.74±1.2341.56±0.903.8700.005Ca2+-ATP酶 1周38.26±0.3541.54±0.404.3870.692 4周25.72±0.2133.68±0.314.4830.025 8周13.85±0.1421.69±0.254.6730.006
3 讨 论
周围神经损伤后骨骼肌丧失神经冲动的传入和支配,由于靶细胞缺乏神经营养因子而发生肌萎缩。NT-3是较重要的神经营养因子,能够维持体外培养的运动神经元的存活,并促进轴突和神经元髓鞘生长,维持肌梭、腱和皮肤传入感觉神经元的存活,促进神经-肌突触的成熟,防止神经损伤后肌肉萎缩[3-4]。有研究发现周围神经损伤后6~12 h内其NT-3水平均下调,2周后NT-3组恢复到对照组水平8倍;经给予外源性NT-3治疗后神经损伤后骨骼肌萎缩有明显的延缓作用,并对轴突再生和神经修复亦具有促进作用[5-6]。但具体作用机制目前研究较少,尚不清楚。
肌细胞凋亡是一种不可逆性的程序性死亡,受细胞内钙超载、自由基、微循环障碍等调节,又是凋亡相关基因caspase、Bcl-2、p53等表达的结果[7]。实验表明NT-3特异性的受体是Trk C ,可通过迅速启动一系列信号转导途径,从而实现对神经元自身信号分子如钙离子通道和神经递质受体的表达和功能状态的调节作用,进一步促进Ca2+-ATP酶发挥作用预防失神经骨骼肌萎缩[8-9]。Ca2+-ATP酶是参与调节Ca2+的主要蛋白,其可以使肌质网内和胞质内 Ca2+保持高浓度差,这是骨骼肌收缩和舒张的基础[10]。周围神经损伤后引起骨骼肌内的Ca2+-ATP酶能量不足,摄取Ca2+能力下降,细胞内和线粒体内Ca2+的大量积聚、钙超载可能激活细胞凋亡的启动程序造成肌细胞凋亡[11]。
caspase是引起细胞凋亡的直接效应物,可以特异地水解底物蛋白,引起染色体凝聚,最终导致细胞凋亡。其中caspase-3是凋亡过程中最重要的蛋白酶,直接介导凋亡实施[12]。研究发现在脊髓损伤后caspase-3表达增加参与了脊髓损伤细胞凋亡的调节,NT-3可能通过抑制caspase-3 基因的转录,减少促凋亡因素,抑制神经细胞的凋亡[13]。
本实验研究表明,caspase-3 基因转录和蛋白表达水平在神经损伤后2 h开始增加,但是NT-3组7 d开始下降,而生理盐水组持续较高水平。应用外源性NT-3 4周后,肌细胞凋亡率较生理盐水组明显减低;失神经后4周腓肠肌内 Ca2+-ATP酶明显下降,NT-3组中Ca2+-ATP酶较生理盐水组明显增加,失神经骨骼肌的收缩功能明显增加(P<0.05)。这说明神经损伤后caspase-3基因表达较高、抗凋亡因素不足和Ca2+-ATP酶下降是导致肌细胞凋亡的原因之一,而给予NT-3干预后骨骼肌细胞的增殖能力增强并能下调活化caspase-3基因和蛋白表达和升高Ca2+-ATP酶,减少促凋亡因素,抑制肌细胞的凋亡,从而减缓和保护失神经后肌萎缩促进神经再生恢复,为脑、脊髓中枢和周围神经损伤后的修复提供理论依据。
参考文献:
[1] Kato K,Yamanouchi D,Esbona K,et al.Caspase-mediated protein kinase C-delta cleavage is necessary for apoptosis of vascular smooth muscle cells[J].Am J Physiol Heart Circ Physiol,2009,297(6):2253-2261.
[2] Hagg T,Baker KA,Emsley JG,et al.Prolonged local neurotrophin-3 infusion reduces ipsilateral collateral sprouting of spared corticospinal axons in adult rats[J].Neuroscience,2005,130(4):875-887.
[3] Kolpakov MA,Seqqat R,Rafiq K,et al.Pleiotropic effects of neutrophils on myocyte apoptosis and left ventricular remodeling during early volume overload[J].J Mol Cell Cardiol,2009,47(5):634-645.
[4] 罗云,黎杏群,唐涛,等.脑溢安对脑出血大鼠脑内神经营养因子-3表达的影响[J].重庆医学,2009,38(24):3105-3108,3196.
[5] Taylor L,Jones L,Tuszynski MH,et al.Neurotrophin-3 gradients established by lentiviral gene delivery promote short-distance axonal bridging beyond cellular grafts in the injured spinal cord[J].J Neurosci,2006,26(38):9713-9721.
[6] Chen Q,Zhou L,Shine HD.Expression of neurotrophin-3 promotes axonal plasticity in the acute but not chronic injured spinal cord[J].J Neurotrauma,2006,23(8):1254-1260.
[7] Song W,Kwak HB,Lawler JM.Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle[J].Antioxid Redox Signal,2006,8(3/4):517-528.
[8] Ying Z,Roy RR,Edgerton VR,et al.Voluntary exercise increases neurotrophin-3 and its receptor TrkC in the spinal cord[J].Brian Res,2003,987(1):93-99.
[9] Radak Z,Chung HY,Goto S.Systemic adaptation to oxidative challenge induced by regular exercise[J].Free Radic Biol Med,2008,44(2):153-159.
[10] Wang W,Zhang H,Gao H,et al.beta 1-Adrenergic receptor activation induces mouse cardiac myocyte death through both L-type calcium channel-dependent and -independent pathways[J].Am J Physiol Heart Circ Physiol,2010,299(2):322-331.
[11] Suzuki S,Yokoyama U,Abe T,et al.Differential roles of Epac in regulating cell death in neuronal and myocardial cells[J].J Biol Chem,2010,285(31):24248-24259.
[12] Perier C,Bové J,Dehay B,et al.Apoptosis-inducing factor deficiency sensitizes dopaminergic neurons to parkinsonian neurotoxins[J].Ann Neurol,2010,68(2):184-192.
[13] Yune TY,Kim SJ,Lee SM,et al.Systemic administration of 17beta-estradiol reduces apoptotic cell death and improves functional recovery following traumatic spinal cord injury in rats[J].J Neurotrauma,2004,21(3):293-306.
猜你喜欢
赣南医学院学报(2019年6期)2019-07-26
中成药(2018年10期)2018-10-26
现代电生理学杂志(2016年1期)2016-07-10
中西医结合心脑血管病杂志(2016年20期)2016-03-01
磁共振成像(2015年5期)2015-12-23
中国康复理论与实践(2015年7期)2015-05-09
中国当代医药(2015年33期)2015-03-01
西安交通大学学报(医学版)(2015年2期)2015-02-28
中华骨与关节外科杂志(2014年4期)2014-01-22
中国病理生理杂志(2012年8期)2012-03-17
