
Henry反应在手性药物不对称合成中的应用
2014-03-14 05:14刘丹丹王耀先赵干卿
精细石油化工 2014年6期
刘丹丹,王耀先, 赵干卿
(平顶山学院,河南 平顶山 467002)
Henry反应又称Nitro-Aldol反应,是由Henry等[1]于1895年发现的,是指在碱催化下硝基烷烃对醛或者酮的羰基进行1,2-加成反应,可以生成β-硝基醇化合物。官能团富集的硝基醇产物可以通过多种转化得到各种用途的化合物,如通过还原可得到氨基醇,脱硝基可以得到醇,脱水可以得到硝基烯烃,氧化可以得到硝基酮等(见图1)[2-3]。
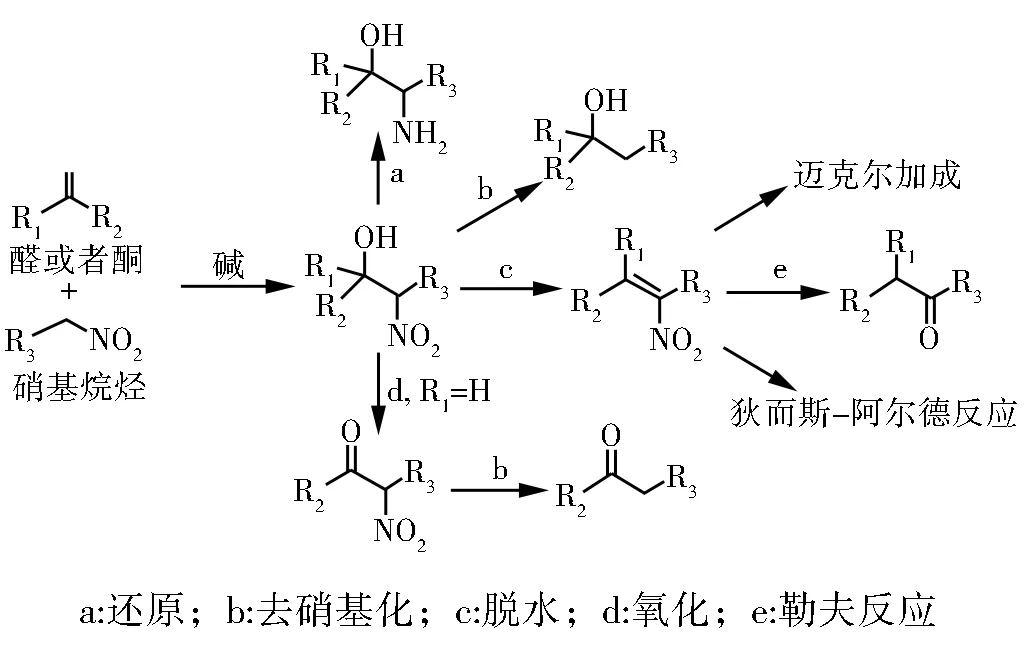
图1 Henry反应以及β-硝基醇产物的各种转化
1992年,Sasai等[4]报道了第一例不对称Henry反应,之后各种类型的催化体系不断出现,获得了很高的产率、对映选择性和非对映选择性[5-8]。我国科学家曾对催化不对称Henry反应做过总结[9-12],但针对其在手性药物合成中的应用未见报道,本文对此进行了阐述。
1 醛的不对称Henry反应
1.1 醛和硝基甲烷的不对称Henry反应
Sasai等[13]将手性联萘酚配合镧锂双金属催化剂A应用到(S)-普蔡洛尔(propranolol)的合成当中,从1-萘基保护的羟基乙醛出发,通过和硝基甲烷的不对称Henry反应,再将硝基还原成氨基,一锅法在丙酮中N-异丙基化。(S)-普蔡洛尔在化疗中作β-受体阻断药,用于治疗心率不齐,心绞痛等,它比其(R)-异构体的活性高98倍,可大大减少药物使用量,减轻肝脏的代谢负荷。随后,按类似的方法,将直接购买的4-羟基吲哚,经过多步反应,制备了一种血管舒张药(S)-心得乐(Propranolol)[14]。
Sasai等又从天然氨基酸L-苯丙氨酸出发,制备了手性的氨基醛,还是用联萘酚配合镧系金属催化其和硝基甲烷反应,得到了HIV蛋白酶抑制剂KNI-227和KNI-272的重要中间体[15](见图2)。
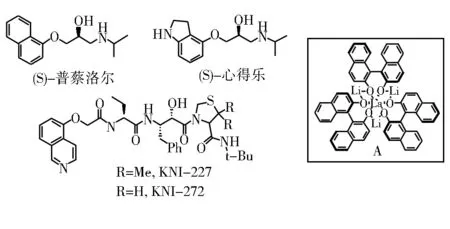
图2 (S)-普蔡洛尔,(S)-心得乐以及
采用类似的不对称Henry反应作为关键步,Corey等[16],使用金鸡纳碱衍生的季铵盐催化剂B也实现了HIV蛋白酶抑制剂安泼那韦的合成。
2002年Trost[17]将基于天然脯氨酸的双核锌催化剂C应用到β肾上腺素受体激动剂药物(-)-地诺帕明和(-)-阿佈他明的合成中,从保护的羟基苯甲醛出发,得到R-型产物,经过后续还原,酰化,还原,脱保护等多步反应,即得到预期产物。
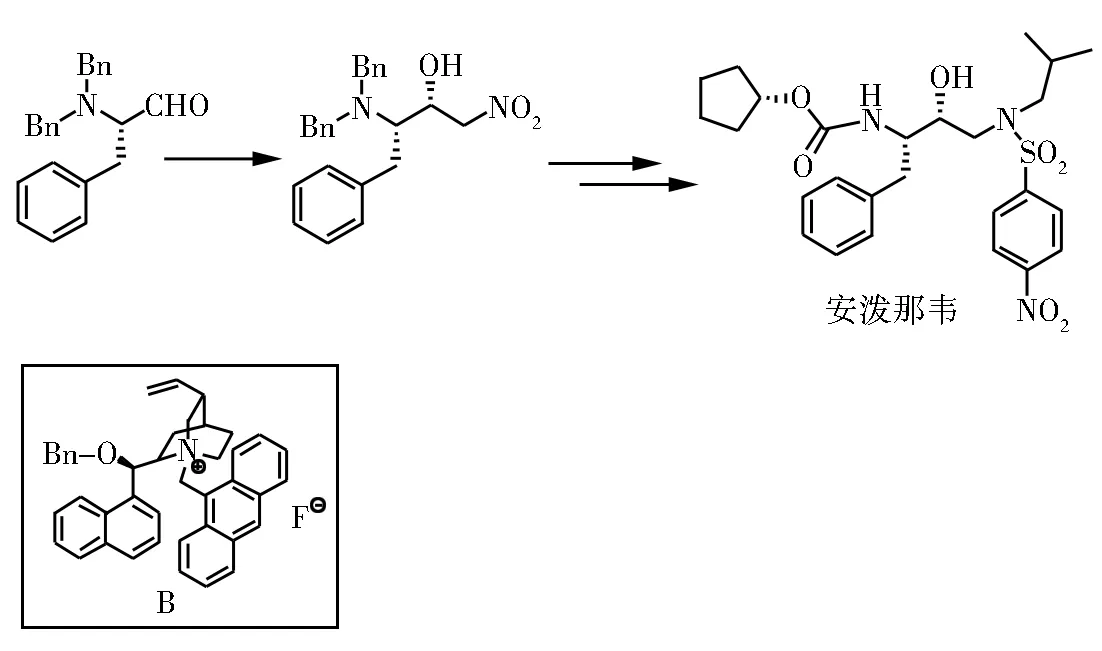
图3 安泼那韦的合成

图4 (-)-地诺帕明和(-)-阿佈他明的合成
四川大学的Feng等[18]用Salen还原亚胺得到的二仲胺Salan配体D配合三氟甲基磺酸亚铜,直接催化3-羟基苯甲醛和硝基甲烷反应,得到硝基醇后再钯碳加氢还原制备得到了(S)-去甲苯福林(图5)。
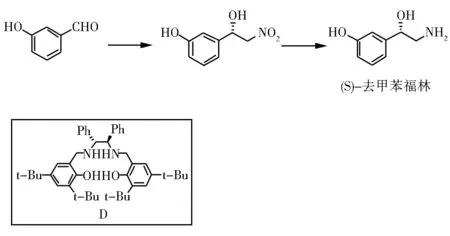
图5 (S)-去甲苯福林的合成
中科大Wang等[19]从4-羟基苯甲醛直接出发,在脯氨酸衍生的手性配体E的促进下,实现了水相中硝基甲烷反应,然后再催化氢化得到 (S)-章鱼胺,再和阿魏酸缩合,得到 (S)-阿魏酰基章鱼胺(图6)。
Savoia等[20]将大环配体用于催化4-Boc保护的羟基苯甲醛和硝基甲烷反应,然后还原得到氨基醇,再和丙酮还原胺化,最后用氯化氢甲醇溶液脱Boc保护,同时氨基成盐,得到 (R)-N-异丙基甲新福林盐酸盐。
White等[21]发展了一种桥环Salan配体催化的Henry反应,成功应用到β受体阻断药,抗高血压药:(S)-托利洛尔,(S)-莫普洛尔以及(S)-普蔡洛尔的合成中。
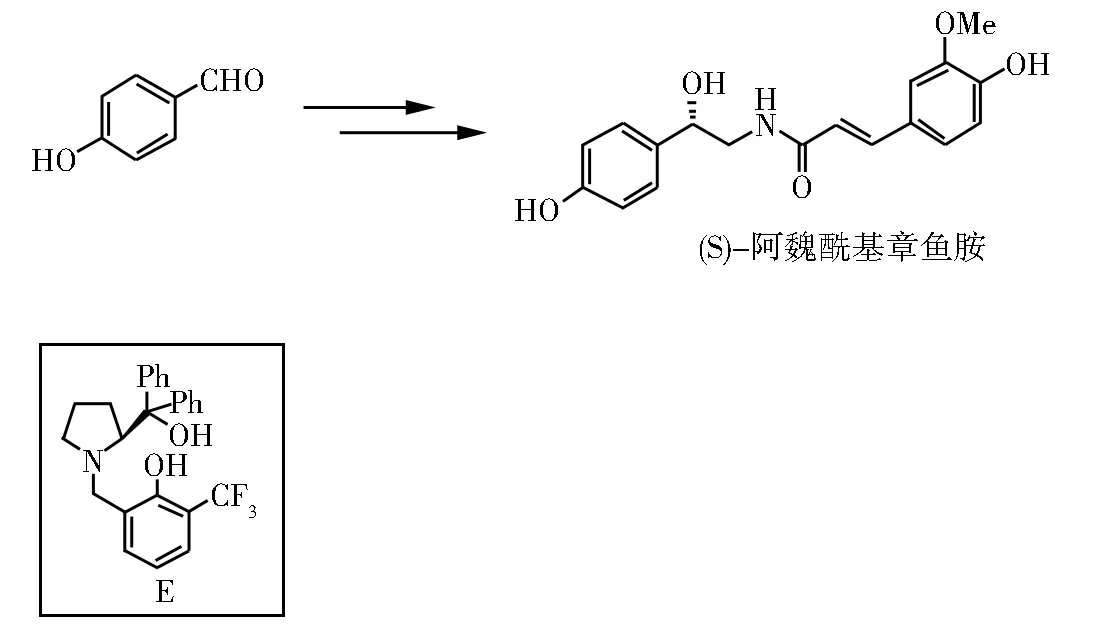
图6 (S)-阿魏酰基章鱼胺的合成
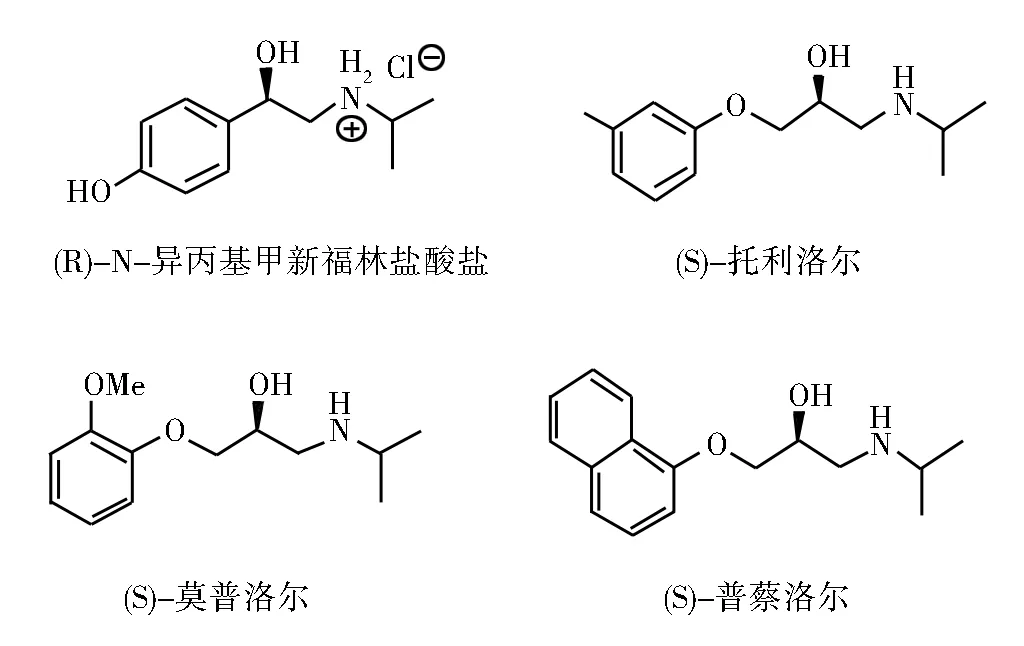
图7 异丙基甲新福林盐酸盐,(S)-托利洛尔,(S)-莫普洛尔以及 (S)-普蔡洛尔的合成
Lu等[22]将鹰爪豆碱铜盐配合物催化的不对称Henry反应成功应用到 (R)-沙美特罗(一种β2促效剂、扩张支气管作用,可以控制支气管哮喘发作症状) 的全合成中。
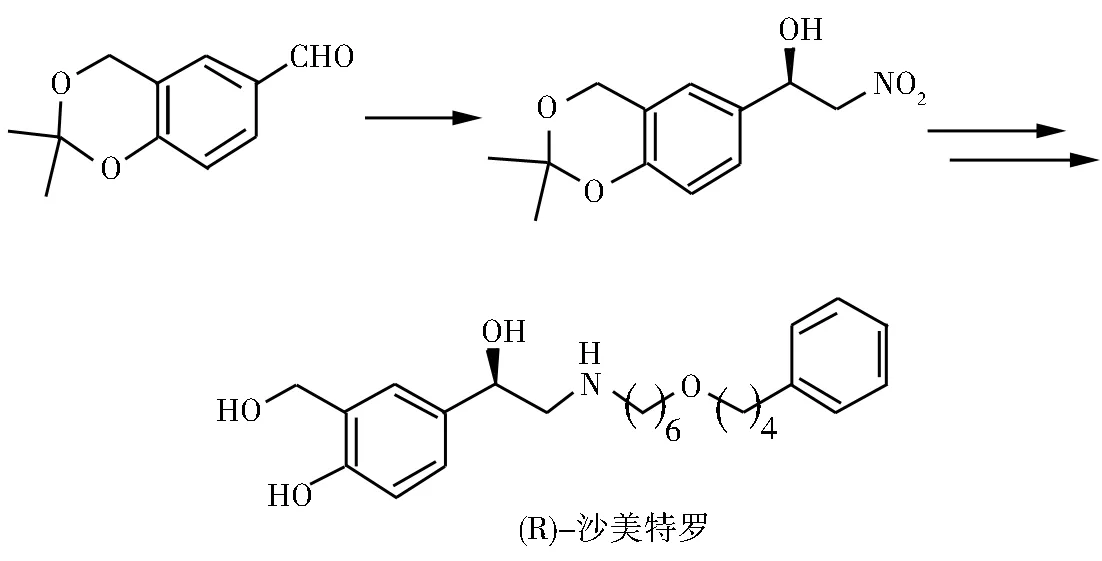
图8 (R)-沙美特罗的合成
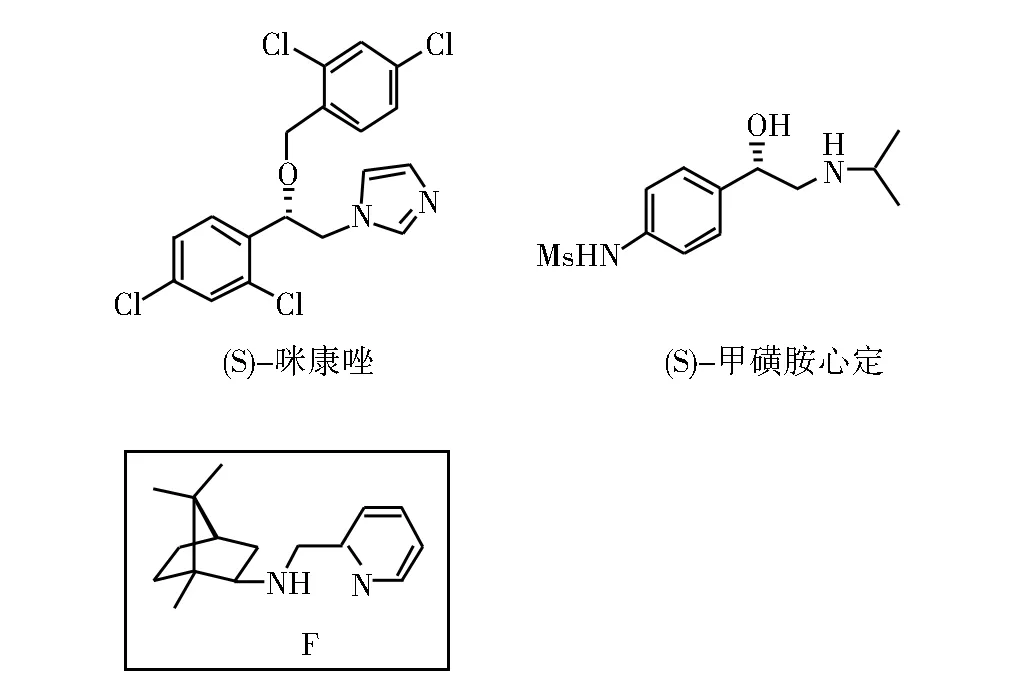
图9 (S)-咪康唑和(S)-甲磺胺心定的合成
2008年,Blay等[23]开发出一类氨基吡啶樟脑配体F,用于催化2,4-二氯苯甲醛和硝基甲烷反应,合成了抗菌药 (S)-咪康唑。Blay等[24]又从对甲磺酰胺基苯甲醛出发,和硝基甲烷反应合成得到β受体阻滞药(S)-甲磺胺心定。
华中科技大学的Gong等[25]将L-脯氨酸和天然樟脑进行组合,开发出一类手性二仲胺的配体G,将其应用到铜催化不对称Henry反应当中,来制备合成(S)-肾上腺素和去甲肾上腺素的重要中间体(S)-2-氨基-1-(3,4-二甲氧基苯基)乙醇。
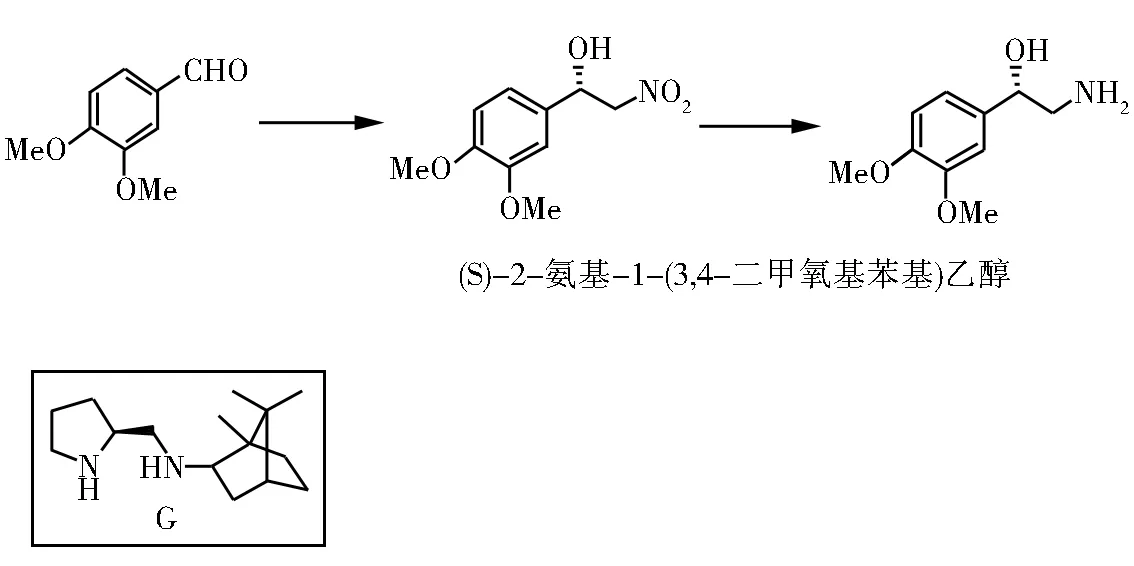
图10 (S)-2-氨基-1-(3,4-二甲氧基苯基)乙醇的合成
1.2 醛和硝基乙烷的不对称Henry反应
除了最常见的硝基甲烷外,硝基乙烷的Henry反应也逐渐被关注。由于其产物具有两个手性中心,所以反应过程中对映选择性和非对映选择性都需要很好的控制,才能得到单一的产物。
Shibasaki等一直致力于双(多)金属中心催化剂的设计合成以及在不对称催化反应中的应用。2008年他们改进了传统的Salen配体,在水杨醛苯环上多引入一个酚羟基,研制了第二代双金属催化剂H。配合金属钯和镧,成功实现了硝基乙烷的高anti选择性Henry反应,并且应用到 β2-肾上腺素抑制剂,利托君(羟苄羟麻黄碱)盐酸盐和β3-肾上腺素抑制剂的合成当中[26]。之后他们又开发了第三代双金属催化剂I。这是基于氨基酸和含氟氨基苯酚以及含氟羟基苯甲酸的二酰胺类配体,配合钕和钠,实现了多相催化硝基乙烷的高anti选择性Henry反应,反应选择性得到提升,ee值和dr值都大幅提高。催化剂可以简单离心沉淀,经过洗涤回收使用。用1%(摩尔分数)的催化剂即可催化50 g的醛和硝基乙烷反应合成 β3-肾上腺素抑制剂[27]。
最近,他们又将第三代催化体系用到默克公司的临床三期降胆固醇药物Anacetrapib的全合成当中[28]。
Wang等[29]从正十六烷基醛出发,直接和硝基乙烷反应,非常简便地合成了从大西洋海哈 (Spisula polynyma) 中提取的一种具有抗肿瘤活性药物 (+)-Spisulosine ES285。
Ooi等[30]从天然缬氨酸出发,制备了季鏻盐催化剂J,可以有效催化硝基乙烷的高anti选择性Henry反应。然后他们把醛底物拓展到炔醛,成功合成了一种从斐济海绵体Xestospongia的代谢物中提取的逆转录酶抑制剂,同时具有广谱抗寄生虫活性和抗菌活性的药物(2S,3R)-(+)-Xestoaminol C和首个不影响中枢神经的抗生素药物(-)-2-epi-Codonopsinine和(-)-Codonopsinine。
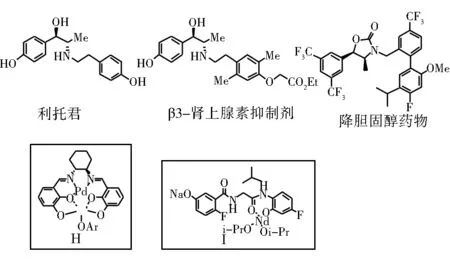
图11 (-)-利托君,β3-肾上腺素抑制剂以及

图12 (+)-Spisulosine ES285的合成

图13 (+)-Xestoaminol C,(-)-2-epi-Codonopsinine和(-)-Codonopsinine的合成
1.3 醛和其他硝基烷烃的不对称Henry反应
除了上述商业上可以购买的硝基烷烃,人们还研究一些官能团化的硝基烷烃的不对称Henry反应,并应用于药物分子的合成当中。
Shibasaki等于1992年最早报道第一代联萘酚镧锂催化不对称Henry反应之后,又对催化剂进行改进,在萘上引入取代三乙基硅炔基,用于催化硝基乙醇的反应,获得高的对映选择性和非对映选择性,并成功应用到苏式-二氢(神经)鞘胺醇 (Threo-dihydrosphingosine) 的合成当中[31]。
2012年,他们又改变了第三代催化剂中氨基酸的构型,用于催化4-硝基1-丁烯的anti选择性Henry反应,合成治疗流行性感冒的药扎那米韦[32],反应过程中有少量的1,4-加成副产物生成。

图14 苏式-二氢(神经)鞘胺醇和扎那米韦的合成
Barua等[33]利用Shibasaki第一代联萘酚配合镧系金属催化剂催化苄基保护的羟基乙醛和硝基苄的直接不对称Henry反应,用于重要抗癌药紫杉醇侧链的合成。

图15 紫杉醇侧链的合成
2012年,Johnston等[34]使用Evens的Box配体催化3,5-二氟苯甲醛和溴代硝基甲烷的不对称Henry反应,经过多步转化,成功实现了可以治疗阿尔茨海默氏病的γ-分泌酶的有效抑制剂LY411575的合成。
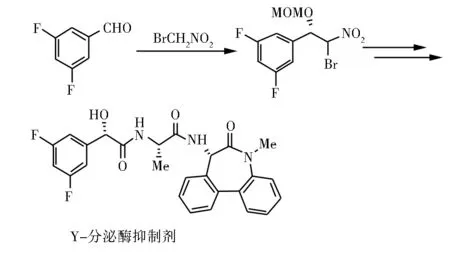
图16 LY411575的合成
2 其他底物的不对称Henry反应
2.1 酮酸酯的不对称Henry反应
2006年,Deng等[35]开发了一类基于天然金鸡纳碱的有机小分子催化剂K,成功实现了有机催化α-酮酸酯和硝基甲烷的不对称Henry反应。进一步将产物进行衍生化,可以得到α-甲基半胱氨酸化合物,这是治疗HIV感染的先导天然化合物(mirabazoles 和thiangazole)的重要中间体。
最近,Cossy等[36-37]用Deng的催化体系,催化了3,4-二氯苯基酮酸酯和硝基甲烷的不对称Henry反应,并用于一种可以治疗精神分裂症的药物神经激肽的拮抗剂受体SSR 241586的合成。
2.2 酮酰胺(靛红)的不对称Henry反应
新墨西哥大学Wang等[38]开发了一种小分子L用来催化靛红和硝基甲烷的不对称Henry反应,并且经过转化成功合成了植物抗菌素(S)-(-)-Spirobrassinin (Scheme 18)。
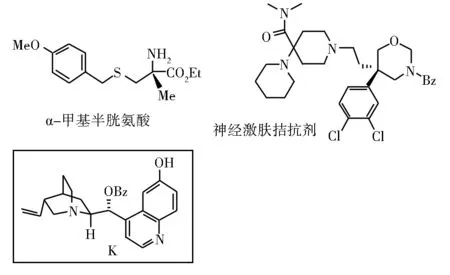
图17 α-甲基半胱氨酸化合物和SSR 241586的合成

图18 (S)-(-)-Spirobrassinin的合成
2.3 亚胺的不对称杂-Henry反应
羰基化合物和伯胺缩合脱水即可合成亚胺化合物,由于其性质和羰基较为相近,是一类非常好的亲电试剂。亚胺和硝基烷烃的加成即为杂-Henry反应, 其产物经过还原可以得到手性邻二胺化合物[39-40]。
日本的Takemoto等[41]开发了一类基于环己二胺的叔胺硫脲类双功能有机小分子催化剂,可以有效促进Boc保护的亚胺和硝基烷烃的不对称杂-Henry反应,并且成功应用到神经激肽受体拮抗剂(+)-CP-99,994的合成中。
Johnston等[42]从亚胺和硝基苄的不对称杂-Henry反应出发,经过多步转化得到小分子抗癌新药p53-MDM2的有效抑制剂(-)-Nutlin-3。
Wang等[43]则从三氟甲基环状酮亚胺出发,和硝基烷烃反应,经过多步转化合成得到了抗HIV药物DPC 083。
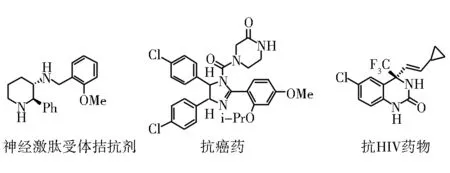
图19 (+)-CP-99,994, (-)-Nutlin-3以及DPC 083的合成
3 结 语
由于不对称Henry用途广泛,近年来,此领域吸引了国内外众多化学家的关注。各种催化体系,包括金属和有机小分子,都得以建立,有性催化剂还可用于手性药物分子的合成,取得了较好的结果。未来不对称Henry反应应用于手性药物的工业化生产,将会是不对称药物合成的研究热点。
参 考 文 献
[1] Henry L. Nitro-alcohols Compt Rend Hebd Séances Acad. Sci. 1895, 120:1265-1267.
[2] Luzzio F A. The Henry Reaction: Recent Examples. Tetrahedron 2001, 57:915-945.
[3] Ono N. The Nitro Group in Organic Synthesis. New York:Wiley-VCH, 2001.
[4] Sasai H, Suzuki T, Shibasaki M, et al. Basic Character of Rare Earth Metal Alkoxides. Utilization in Catalytic C-C Bond-Forming Reactions and Catalytic Asymmetric Nitroaldol Reactions. J Am Chem Soc,1992, 114:4418-4420.
[5] Palomo C, Oiarbide M, Mielgo A. Unveiling Reliable Catalysts for the Asymmetric Nitroaldol (Henry) Reaction. Angew. Chem Int Ed, 2004, 43:5442-5444.
[6] Boruwa J, Gogoi N, Barua N C, et al. Catalytic Asymmetric Henry Reaction. Tetrahedron: Asymmetry, 2006, 17: 3315-3326.
[7] Palomo C, Oiarbide M, Laso A. Recent Advances in the Catalytic Asymmetric Nitroaldol (Henry) Reaction. Eur J Org Chem, 2007:2561-2574.
[8] Blay G, Hernández-Olmos V, Pedro J R. Development of NewN,N-Ligands for the Enantioselective Copper(II)-Catalyzed Henry Reaction. Synlett, 2011, 9: 1195-1211.
[9] 甘昌胜, 潘见. 不对称催化Henry反应研究进展. 有机化学, 2008, 28:1193-1198.
[10] 倪航, 宋庆宝. 铜催化的不对称Henry反应研究进展. 广州化工, 2010, 38: 37-41.
[11] 付记亚,徐小英,王文, 等. 有机小分子催化不对称Henry反应的研究进展. 合成化学, 2010, 18: 1-5.
[12] 高书涛, 郗国宏, 马晶军, 等. 有机催化不对称Henry加成反应. 有机化学, 2010, 30: 1811-1819.
[13] Sasai H, Etoh N, Shibasaki M, et al. Catalytic Asymmetric Nitroaldol Reaction: An Efficient Synthesis of (s) Propranolol Using the Lanthanum Binaphthol Complex. Tetrahedron: Lett, 1993, 34: 855-858.
[14] Sasai H, Suzuki T, Shibasaki M, et al. Syntheses of (S)-(-)-Pindolol and (R)-(-)-Pindolol Utilizing a Lanthanum-Lithium-(R)-BINOL ((R)-LLB) Catalyzed Nitroaldol Reaction. Tetrahedron,1994, 50:12313-12318.
[15] Sasai H, Suzuki T, Shibasaki M, et al. Reaction Utilizing Rare Earth-Li-BINOL Complex. A Highly Efficient Synthesis of Norstatine. Tetrahedron: Lett, 1994, 35: 6123-6126.
[16] Corey E J, Zhang F Y. re- and si-Face-Selective Nitroaldol Reactions Catalyzed by a Rigid Chiral Quaternary Ammonium Salt: A Highly Stereoselective Synthesis of the HIV Protease Inhibitor Amprenavir (Vertex 478). Angew Chem Int Ed, 1999, 38:1931-1934.
[17] Trost B M, Yeh V S C, Ito H, et al. Effect of Ligand Structure on the Zinc-Catalyzed Henry Reaction. Asymmetric Syntheses of (-)-Denopamine and (-)-Arbutamine. Org Lett, 2002, 4:2621-2623.
[18] Xiong Y, Wang F, Feng X, et al. A New Copper(I)-Tetrahydrosalen-Catalyzed Asymmetric Henry Reaction and Its Extension to the Synthesis of (S)-Norphenylephrine. Chem Eur J, 2007, 13, 829-833.
[19] Lai G, Guo F, Wang Z, et al. Highly Enantioselective Henry Reactions in Water Catalyzed by a Copper Tertiary Amine Complex and Applied in the Synthesis of (S)-N-trans-Feruloyl Octopamine. Chem Eur J, 2011, 17:1114-1117.
[20] Gualandi A, Cerisoli L, Savoia D, et al. Pyrrole Macrocyclic Ligands for Cu-Catalyzed Asymmetric Henry Reactions. J Org Chem, 2011, 76, 3399-3408.
[21] White J D, Shaw S. A New Catalyst for the Asymmetric Henry Reaction: Synthesis of β-Nitroethanols in High Enantiomeric Excess. Org Lett,2012, 14:6270-6273.
[22] Guo Z, Deng Y, Lu G, et al. Enantioselective Synthesis of (R)-salmeterol Employing an Asymmetric Henry Reaction as the Key Step. Tetrahedron: Asymmetry, 2011, 22:1395-1399.
[23] Blay G, Domingo L R, Pedro J R, et al. New Highly Asymmetric Henry Reaction Catalyzed by Cu(II) and a C1-Symmetric Aminopyridine Ligand, and Its Application to the Synthesis of Miconazole. Chem Eur J,2008, 14:4725-4730.
[24] Blay G, Hernández-Olmos V, Pedro J,R. Synthesis of (S)-(+)-sotalol and (R)-(-)-isoproterenol via a catalytic enantioselective Henry reaction. Tetrahedron: Asymmetry,2010, 21: 578-581.
[25] Zhou Y, Dong J, Gong Y, et al. Synthesis of C1-Symmetric Chiral Secondary Diamines and Their Applications in the Asymmetric Copper(Ⅱ)-Catalyzed Henry (Nitroaldol) Reactions. J Org Chem, 2011, 76, 588-600.
[26] Handa S, Matsunaga S, Shibasaki M, et al. A Heterobimetallic Pd/La/Schiff Base Complex for anti-Selective Catalytic Asymmetric Nitroaldol Reactions and Applications to Short Syntheses of β-Adrenoceptor Agonists. Angew. Chem Int Ed, 2008, 47: 3230-3233.
[27] Nitabaru T, Kumagai N, Shibasaki M, et al. anti-Selective Catalytic Asymmetric Nitroaldol Reaction via a Heterobimetallic Heterogeneous Catalyst. J Am Chem Soc, 2009, 131:13860-13869.
[28] Ogawa T, Kumagai N, Shibasaki M. Self-Assembling Neodymium/Sodium Heterobimetallic Asymmetric Catalyst Confined in a Carbon Nanotube Network. Angew Chem Int Ed, 2013, 52:6196-6201.
[29] Xu K, Lai G, Wang Z, et al. A Highly anti-Selective Asymmetric Henry Reaction Catalyzed by a Chiral Copper Complex: Applications to the Syntheses of (+)-Spisulosine and a Pyrroloisoquinoline Derivative. Chem Eur J, 2012, 18:12357-12362.
[30] Uraguchi D, Nakamura S, Ooi T. Catalytic Asymmetric Direct Henry Reaction of Ynals: Short Syntheses of (2S,3R)-(+)-Xestoaminol C and (-)-Codonopsinines. Angew Chem Int Ed, 2010, 49:7562-7565.
[31] Sasai H, Tokunaga T, Shibasaki M, et al. Efficient Diastereoselective and Enantioselective Nitroaldol Reactions from Prochiral Starting Materials: Utilization of La-Li-6,6′-Disubstituted BINOL Complexes as Asymmetric Catalysts. J Org Chem, 1995, 60:7388-7389.
[32] Nitabaru T, Kumagai N, Shibasaki M. Catalytic Asymmetric anti-Selective Nitroaldol Reaction En Route to Zanamivir. Angew Chem Int Ed, 2012, 51:1644-1647.
[33] Borah J C, Gogoi S, Barua N C, et al. A Highly Efficient Synthesis of the C-13 Side-chain of Taxol Using Shibasaki’s Asymmetric Henry Reaction. Tetrahedron: Lett, 2004, 45: 3689-3691.
[34] Leighty M W, Shen B, Johnston J N. Enantioselective Synthesis ofαOxy Amides via Umpolung Amide Synthesis. J Am Chem Soc, 2012, 134:15233-15236.
[35] Li H, Wang B, Deng L. Enantioselective Nitroaldol Reaction of r-Ketoesters Catalyzed by Cinchona Alkaloids. J Am Chem Soc, 2006, 128:732-733.
[36] Cochi A, Métro T, Cossy J, et al. Enantioselective Synthesis of SSR 241586 by Using an Organo-Catalyzed Henry Reaction. Org Lett, 2010, 12:3693-3695.
[37] Métro T, Cochi A, Cossy J, et al. Asymmetric Synthesis of an Antagonist of Neurokinin Receptors: SSR 241586. J Org Chem, 2011, 76: 2594-2602.
[38] Liu L, Zhang S, Wang W, et al. Catalytic Enantioselective Henry Reactions of Isatins: Application in the Concise Synthesis of (S)-(-)-Spirobrassinin. Chem Eur J. 2011, 17: 7791-7795.
[38] Marqués-López E, Merino P, Herrera R P, et al. Catalytic Enantioselective Aza-Henry Reactions. Eur J Org Chem, 2009, 2401-2420.
[40] Noble A, Anderson J C. Nitro-Mannich Reaction. Chem Rev, 2013, 113: 2887-2939.
[41] Xu X, Furukawa T, Takemoto Y, et al. Bifunctional-Thiourea-Catalyzed Diastereo- and Enantioselective Aza-Henry Reaction. Chem Eur J, 2006, 12:466-476.
[42] Davis T A, Johnston J N. Catalytic, Enantioselective Synthesis of Stilbene cis-Diamines: A Concise Preparation of (-)-Nutlin-3, a Potent p53/MDM2 Inhibitor. Chem Sci, 2011, 2:1076-1079.
[43] Xie H, Zhang Y, Wang W, et al. Bifunctional Cinchona Alkaloid Thiourea Catalyzed Highly Efficient, Enantioselective Aza-Henry Reaction of Cyclic Trifluoromethyl Ketimines: Synthesis of Anti-HIV Drug DPC 083. Angew Chem Int Ed, 2011, 50:11773-11776.
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13
化工管理(2021年7期)2021-05-13
材料科学与工程学报(2016年4期)2017-01-15
中国农资(2016年1期)2016-12-01
合成化学(2015年4期)2016-01-17
化工进展(2015年3期)2015-11-11
无机化学学报(2014年8期)2014-02-28
无机化学学报(2014年6期)2014-02-28
无机化学学报(2014年5期)2014-02-28
江西理工大学学报(2013年1期)2013-03-20
