
天蓝色链霉菌代谢物组测定方法优化及其代谢特征
2014-03-10 01:46李宜鸿李珊珊艾国民王为善张部昌杨克迁
生物工程学报 2014年4期
李宜鸿,李珊珊,艾国民,王为善,张部昌,杨克迁
1 安徽大学生命科学学院,安徽 合肥 230601
2 中国科学院微生物研究所 微生物资源前期开发国家重点实验室,北京 100101
链霉菌能够产生众多具有生物活性的次级代谢产物,具有重要的研究与应用价值。其模式生物天蓝色链霉菌Streptomyces coelicolor在2002年就已经完成基因组测序,具有清晰的遗传背景[1]。同时,该菌株能够产生具有颜色的红色次级代谢产物十一烷基灵菌红素 (Undecylprodigiosin,Red) 与蓝色次级代谢产物放线紫红素(Actinhordin,Act)。因此,该菌是研究抗生素生物合成调控和链霉菌代谢工程改造的理想对象。
随着组学技术的发展,系统生物学研究的地位日益显著。代谢物作为生物信息传递的终端层次,在基因功能诠释和代谢状态分析等方面表现出了巨大的潜力。代谢物组学是一种通过定性定量测量生物体代谢产物的种类和浓度变化,分析系列关联代谢物的综合差异,从而发现生物系统对基因以及环境变化响应的研究方法[2-3]。由于代谢物组学能够从系统生物学的高度集中阐释生物体代谢所反映的全部信息,因此越来越多地受到科学家们的重视。代谢物组学在工业微生物领域的应用主要体现在以下几个方面:快速识别微生物在培养过程中的生理与代谢特征[4];发现菌株新的代谢途径[5];从系统水平阐述微生物代谢调控机制[6];理性指导菌株改造、提高靶标代谢物的产量并指导发酵过程优化[7]等。在工业微生物领域,代谢物组学的研究工作主要集中于酵母、大肠杆菌和枯草芽胞杆菌等模式微生物[6-9]。近期,也有部分针对链霉菌的研究工作[9-10]。由于链霉菌可产生丰富的次级代谢产物,而目前根据基因簇所获得的化合物信息仍然有限,因此,研究链霉菌胞外次级代谢物组,可以发现新的次级代谢化合物,并促进对次级代谢合成基因簇的认识[10]。此外,代谢物组学也可以用于研究链霉菌对环境压力的响应机制[11]。这些研究工作表明,对于具有复杂生理代谢的链霉菌而言,代谢物组学这一研究手段可以发挥重要的作用。
代谢物组学研究依赖的技术平台主要有核磁共振 (NMR) ,以及色谱-质谱联用,包括气相色谱-质谱 (GC-MS)、液相色谱-质谱 (LC-MS)和毛细管电泳-质谱 (CE-MS)。其中,由于GC-MS具有灵敏度高、分辨率高和可选择性地分离和检测大量痕量代谢物和同质异构体等优点,成为微生物代谢物组学研究中应用最广泛的分析方法[12]。代谢物组学研究方法可分为 3个主要部分:样品制备、代谢物检测和数据分析 (图1) 。其中,获得能够准确反映细胞瞬时状态的代谢物样品是该方法的关键环节。微生物生长条件和状态各异,因此,分离胞内外代谢物方法也有所不同。对于研究较多的大肠杆菌 (革兰氏阴性菌)、枯草芽胞杆菌 (革兰氏阳性菌) 和酵母菌 (真菌) 而言,已有不少研究者对其代谢物组学样品制备方法进行了摸索优化[13-14]。对于链霉菌,目前却仍无针对性的样品制备方法优化报道。由于获得能够准确捕捉代谢物信息的测试样品对研究链霉菌代谢行为及指导抗生素代谢工程具有重要意义,因此有必要对链霉菌样品制备方法进行优化。
本工作以天蓝色链霉菌为研究对象,对其代谢物组样品制备方法进行了系统优化,并利用GC-MS分析平台分析了该菌株的主要代谢特征。此外,本工作还利用优化的样品制备方法比较了天蓝色链霉菌 M145在不同生长阶段各个代谢途径的相对强度,找出了这些代谢途径和次级代谢生物合成的相关性,可以为其他链霉菌的代谢物组分析工作提供参考。
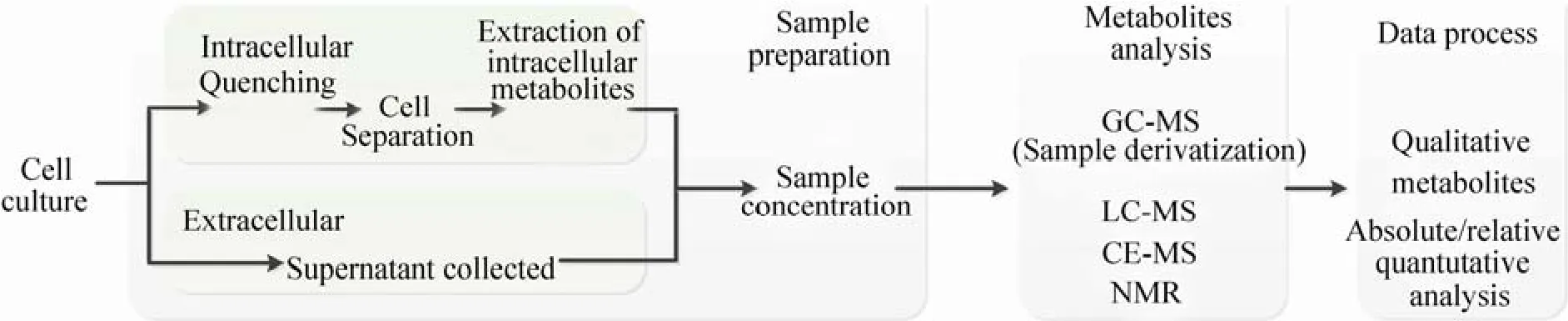
图1 代谢物组学分析流程Fig. 1 Flowchart for a standard protocol of metabolome analysis.
1 材料与方法
1.1 材料
1.1.1 菌株
本实验使用的菌株为天蓝色链霉菌M145。
1.1.2 试剂
衍生化试剂甲氧氨盐酸盐、N-甲基-N- (三甲基硅基) 三氟乙酰胺 (MSTFA),以及内标物琥珀酸-2,2,3,3-d4均购自Sigma公司。甲醇、吡啶和正已烷试剂均为色谱级别。其他试剂均为分析纯级别。
1.1.3 主要仪器
Agilent GC-MS (Agilent 7896A/5975C GC/MS)、高速冷冻离心机 (Eppendorf 5415R)、超低温冰箱 (DW-86L626)、真空冷冻干燥机(FD-1A-50)、真空泵 (GM-0.33a)、酶标仪(Synergy H4 Hybrid Reader)、HPLC (Prominence LC-20AT)。
1.2 培养基及培养条件
1.2.1 培养基
孢子培养基为MS培养基 (g/L):黄豆粉20,甘露醇20,Agar 20,121 ℃灭菌20 min。发酵培养基为R2YE培养基 (g/L):蔗糖103,K2SO40.25,MgCl210.12,葡萄糖10,酸水解酪素0.1,酵母提取物 5,微量元素 (ZnCl20.04,FeCl3·6H2O 0.2,CuCl2·2H2O 0.01,MnCl2·4H2O 0.01,Na2B4O7·10H2O 0.01,(NH4)6Mo7O24·4H2O 0.01) 2 mL,蒸馏水800 mL,灭菌后加入KH2PO41 mL, CaCl2·2H2O 8 mL,L-proline 10 mL,NaOH 0.5 mL,115 ℃灭菌30 min。
1.2.2 培养条件
将冻存于–80 ℃的孢子划线接种于MS培养基,于28 ℃培养4 d,收集新鲜孢子接种于R2YE培养基中,接种浓度为4×108个孢子/100 mL培养基,28 ℃、250 r/min培养6 d。
1.3 样品制备方法
取样后,将 1 mL菌液迅速离心 (4 ℃、13 000×g) 10 min,保留上清,用于测定放线紫红素的产量,收集的菌体用于测定十一烷基灵菌红素的产量。其余样品迅速低温淬灭后,用于制备胞内代谢物样品。
1.3.1 细胞淬灭
将细胞培养物按1:4的比例迅速加入60%(V/V) 甲醇/水溶液中 (–40 ℃预冷 24 h),摇匀后于–40 ℃静置 (按需要分别静置 2、4、6、8和 10 min)。
1.3.2 菌体分离
采用离心与快速过滤两种方法分离菌体。1) 离心法:将细胞淬灭物进行冷冻离心,(–9 ℃,2 000×g,3 min),弃上清并用 0 ℃预冷的去离子水洗涤菌体,重复 3次,最后一次离心条件为–9 ℃,8 000×g,5 min。2) 快速过滤法:利用真空泵对细胞淬灭物进行快速过滤并用0 ℃预冷的去离子水洗涤菌体 (1 min内完成所有步骤)。将两种方法得到的菌体冻存于–80 ℃。
1.3.3 代谢物提取与浓缩
将收集的菌体利用液氮研磨,收集300 mg粉末于2 mL离心管中,加入1 mL 50% (V/V) 甲醇/水溶液 (–40 ℃预冷24 h),剧烈振荡后迅速投入液氮中速冻,设置速冻时间分别为 45 s、3 min和5 min,之后于冰上融化,重复3次。然后将混合物低温离心 (–9 ℃,13 000×g,5 min),取800 μL上清至15 mL离心管中。再向2 mL离心管中加入500 μL预冷的50% (V/V)甲醇/水溶液,剧烈振荡后以相同的条件离心,再将500 μL上清转移至15 mL离心管中,并加入20 μL 0.2 mg/mL D4-琥珀酸作为内标。将抽提的代谢物溶液于–80 ℃过夜冷冻后,冻干24 h。
1.3.4 代谢物衍生化
将冻干样品加入 50 μL甲氧胺盐吡啶溶液(20 mg/mL),于水浴中加热 90 min (37 ℃或40 ℃);之后加入 80 μL MSTFA 继续加热90 min (37 ℃[15]或 40 ℃[16]),得到 GC-MS 分析样品。
1.4 GC-MS代谢物检测方法
色谱条件为:气相色谱分析仪Agilent 7890A,色谱柱 DB-5MS (30 m×0.25 mm,0.25 μm,Agilent);柱温箱升温程序70 ℃保持2 min,然后以5 /min℃升温至290 ℃并保持3 min;进样口及检测器温度均保持为250 ℃;GC-MS接口温度为280 ℃;载气为He气,流速1.0 mL/min;样品进样量为1 μL,分流比为20:1。质谱条件:质谱仪 Aglient 5975C MSD System;电子轰击电离 (EI+源),EI+源温度 250 ℃,轰击能量eV,离子电流40 μA;扫描分子量范围25–650;扫描速度2 scan/second。
1.5 其他分析方法
1.5.1 采用二苯胺法[17]确定生物量
收集1 mL发酵液,离心去上清后用SET 缓冲液洗1次,离心去上清;将菌体重悬于2 mL二苯胺试剂中,60 ℃水浴1 h,之后离心取上清测定OD595。每个样品设置3个生物学重复。
1.5.2 放线紫红素产量测定方法
收集1 mL发酵液,4 ℃、10 000×g离心取上清,加入终浓度1 mol/L的KOH,离心取上清,利用酶标仪测定OD640。根据该波长下的摩尔吸光系数 (ε640=25 320) 计算 Act产量[18]。每个样品设置3个生物学重复。
1.5.3 十一烷基灵菌红素产量测定
收集1 mL发酵液,4 ℃、10 000×g离心收集菌体,1 mol/L HCl洗涤菌体2次,1 mL甲醇萃取,离心取上清,利用酶标仪测定OD530,根据该波长下的摩尔吸光系数 (ε530=100 500) 计算Red产量[19]。每个样品设置3个生物学重复。
1.5.4 数据处理方法
代谢物数据的相对定量是根据总离子流峰进行的。代谢物组分的获得方法是将由 Agilent 7890/5975C MSD System采集的原始数据导入解卷积软件AMDIS中分析得到。将解卷积后的数据与NIST 8.0库、Fiehn代谢物数据库进行比对搜索得到相应的代谢物 (检索匹配度要求高于 80)。进一步结合 METLIN数据库和 KEGG数据库对代谢物所属途径进行分类。将质谱峰面积分别对各样品的生物量和内标物的峰面积进行归一化,获得胞内代谢物的相对浓度。每个样品设置 3个生物学重复。分别选择对数期和稳定期的胞内代谢物样品中检测到的部分代谢物,以及胞外Act产量为例,利用t-test检验样品间的差异显著性。
2 结果
2.1 样品数据的重复性检测
为了获得精确的数据,实验初始首先对GC-MS仪器检测的重复性进行了考察。以内标物D4-琥珀酸为例,分别进样3次,其出峰时间分别为16.160、16.162和16.171 min,这个结果表明仪器的重复性很好。
为了检验不同生物学样品的重复性,本实验取24 h和72 h的3个样品所采集到的数据进行了比较。将胞内代谢物数据进行归一化后,随机抽取不同胞代谢物的总离子流质谱峰面积,以及胞外代谢产物 Act的浓度。如表 1所示,3个生物学样品的P-value均大于0.1,表明样本间的差异不显著,所取的 3个生物学样品的重复性良好,可用于后续数据分析。
2.2 不同样品处理条件对代谢物提取的影响
2.2.1 细胞淬灭时间优化
细胞淬灭能够及时终止胞内的代谢活动,是获得能够准确反映细胞瞬时生理状态的代谢物样品的关键环节。本研究以冷甲醇水为淬灭溶剂,着重研究了淬灭时间对代谢物提取的影响。迅速从同一培养瓶中取5份天蓝色链霉菌M145的对数中期培养物 (30 h),并同时分别与–40 ℃预冷的 60% (V/V) 甲醇水按 1:4体积混合,置于–40 ℃分别静置 2、4、6、8和10 min,然后提取代谢物后进行GC-MS分析。结果如图2所示,淬灭时间为2 min时,提取到的代谢物组分数为 (272±13),当淬灭时间延长至4 min时,提取到的代谢物组分数目最多,达到356个。然而继续延长淬灭时间代谢物组分数则会显著下降,10 min时仅有 (110±8) 个,较4 min时降低69.1%。此外,如图2所示,将总离子流 (TIC) 峰面积设置为105时,淬灭4 min时所获得的代谢物TIC峰面积大于该值的代谢物数目分别较其他 4个时间点多 46%(2 min)、29% (6 min)、68% (8 min) 和 89%(10 min)。这些数据表明4 min是天蓝色链霉菌淬灭效果最好的时间段。
2.2.2 细胞分离条件优化
快速干净地分离菌体与淬灭液和胞外代谢物的混合物,能够减少胞内代谢物损失和外界杂质干扰。对比低温离心与快速抽滤两种方法,结果显示针对同一天蓝色链霉菌M145样品,快速抽滤可得到 (373±9) 个组分峰,较低温离心法提取的组分峰数目 (312±12) 高 19.6%。此外,抽滤法得到的TIC峰面积大于105的代谢物数目较低温离心法高65%。
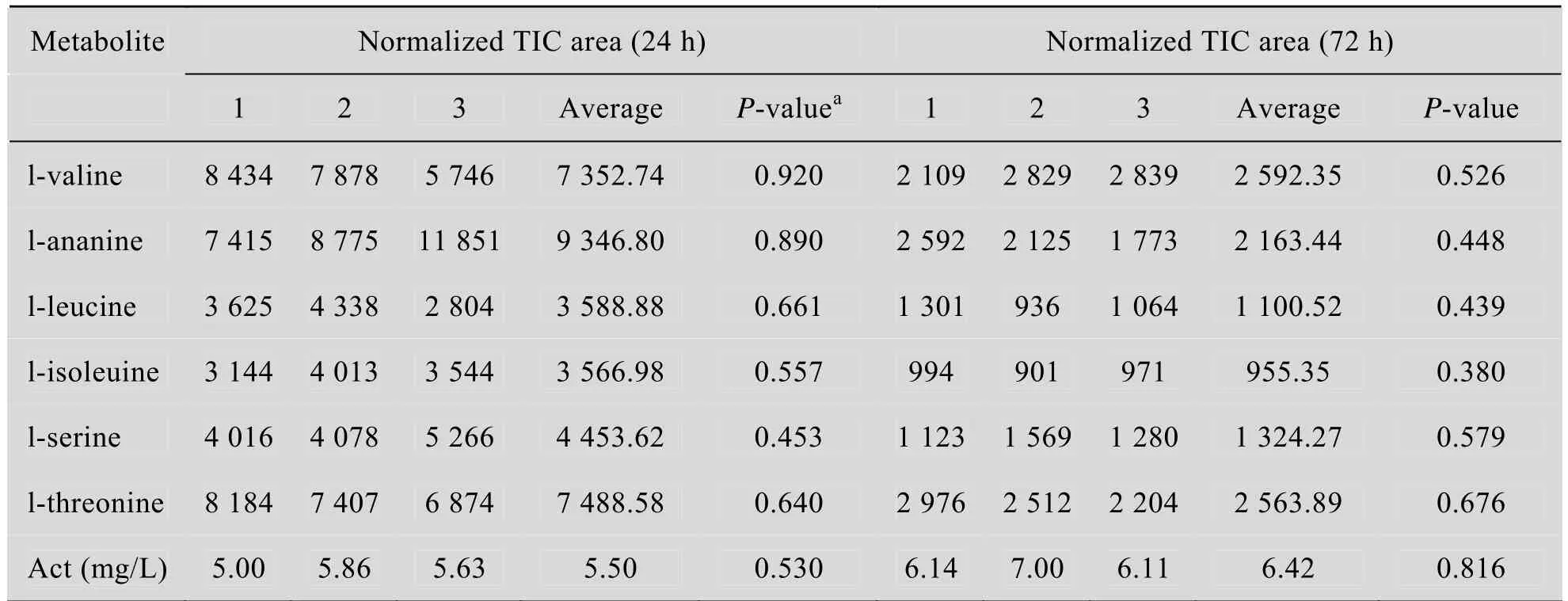
表1 样品生物学重复检验Table 1 Significance test of different biological samples
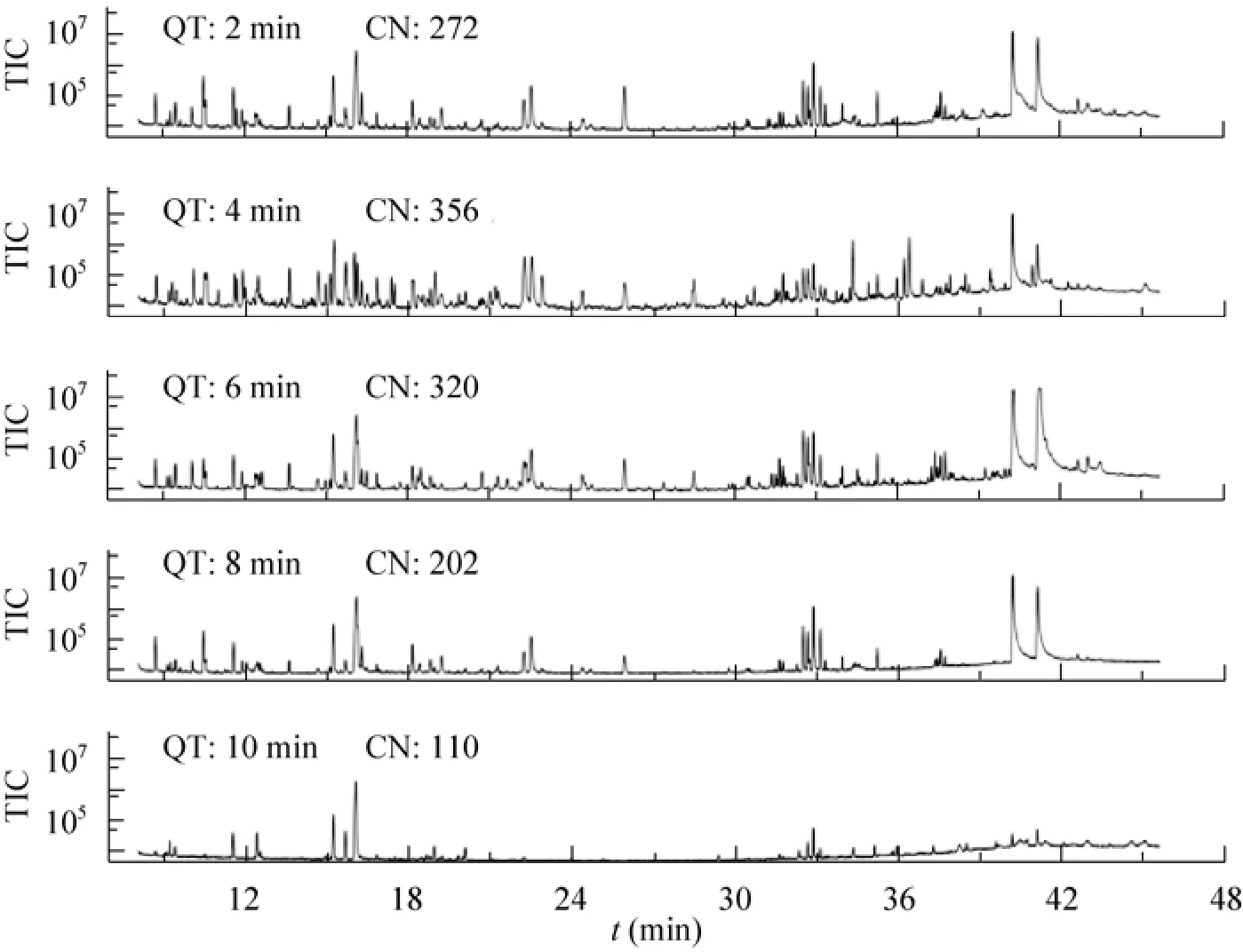
图2 不同淬灭时间对代谢物提取的影响Fig. 2 Impact of quenching times on the extration of metabolites. QT: quenching time; CN: number of metabolite components; TIC: total ion chromatogram.
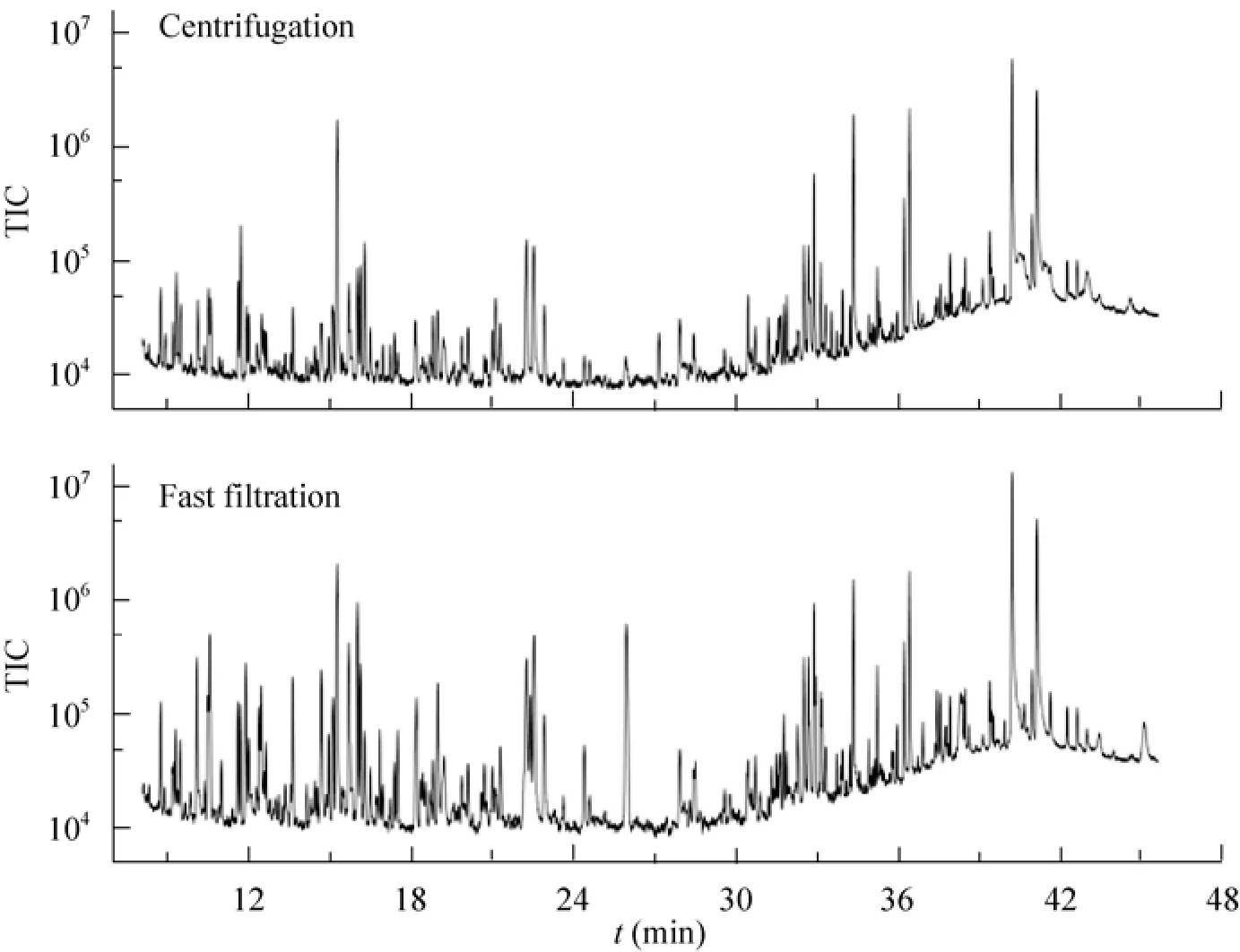
图3 不同菌体分离方式对代谢物提取的影响Fig. 3 Impact of different cell separation methods on metabolite extraction after quenching. TIC: total ion chromatogram.
2.2.3 代谢物提取条件的优化
实验进一步优化了天蓝色链霉菌代谢物提取条件。为了使代谢物充分释放,采用液氮研磨的方式对细胞壁进行破坏,利用–40 ℃预冷的50% (V/V) 甲醇水多次抽提及液氮-冰水反复冻融提取代谢物。实验比较了不同的液氮速冻时间(45 s、3 min和5 min) 对代谢物提取的影响,结果表明45 s的速冻时间能够检测到 (464±17) 种代谢物组分,而延长速冻时间至3 min和5 min则会导致代谢物组分分别较45 s减少24.8%和28.7%。此外,45 s液氮速冻时间获得的代谢物中,TIC峰面积大于105的数目较其他两个时间点分别高27%和59%。这些结果表明在液氮中速冻45 s而后在冰上融化是反复冻融的较好条件。
2.2.4 代谢物衍生化条件的优化
提取获得胞内代谢物后,衍生化过程能够增加代谢物的稳定性并降低其沸点,有利于GC-MS检测胞内代谢物。本工作比较了37 ℃和40 ℃两个温度条件下衍生化试剂的衍生效率,结果显示,37 ℃处理代谢物分别与甲氧胺盐和MSTFA反应90 min,仍有13.7%的代谢物仅被部分衍生化。而在40 ℃处理下,试剂的衍生化效率可达到96.7%。
2.3 GC-MS可检测代谢物在天蓝色链霉菌代谢网络中的覆盖度
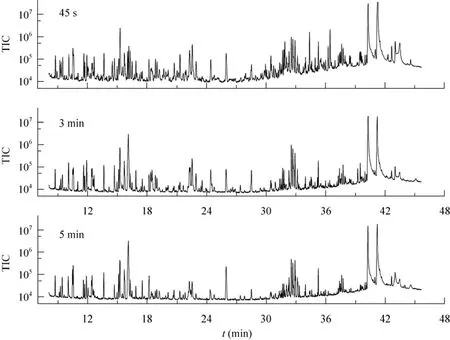
图4 不同液氮速冻时间对代谢提取的影响Fig. 4 Impact of the different freeze-thaw times in liquid nitrogen on metabolite extration. TIC: total ion chromatogram.
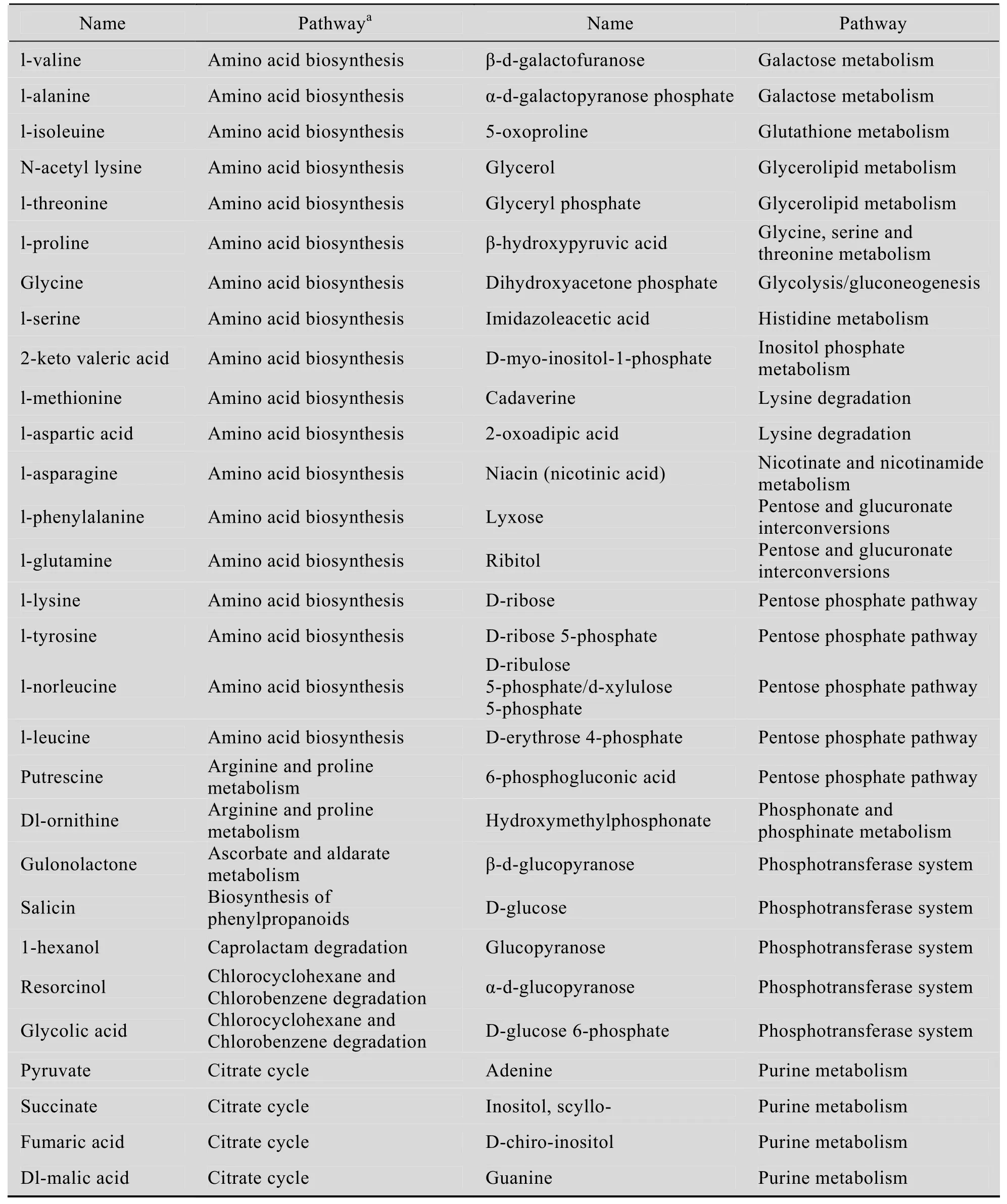
表2 GC-MS检测到的胞内代谢物Table 2 Intracellular metabolites identified by GC-MS
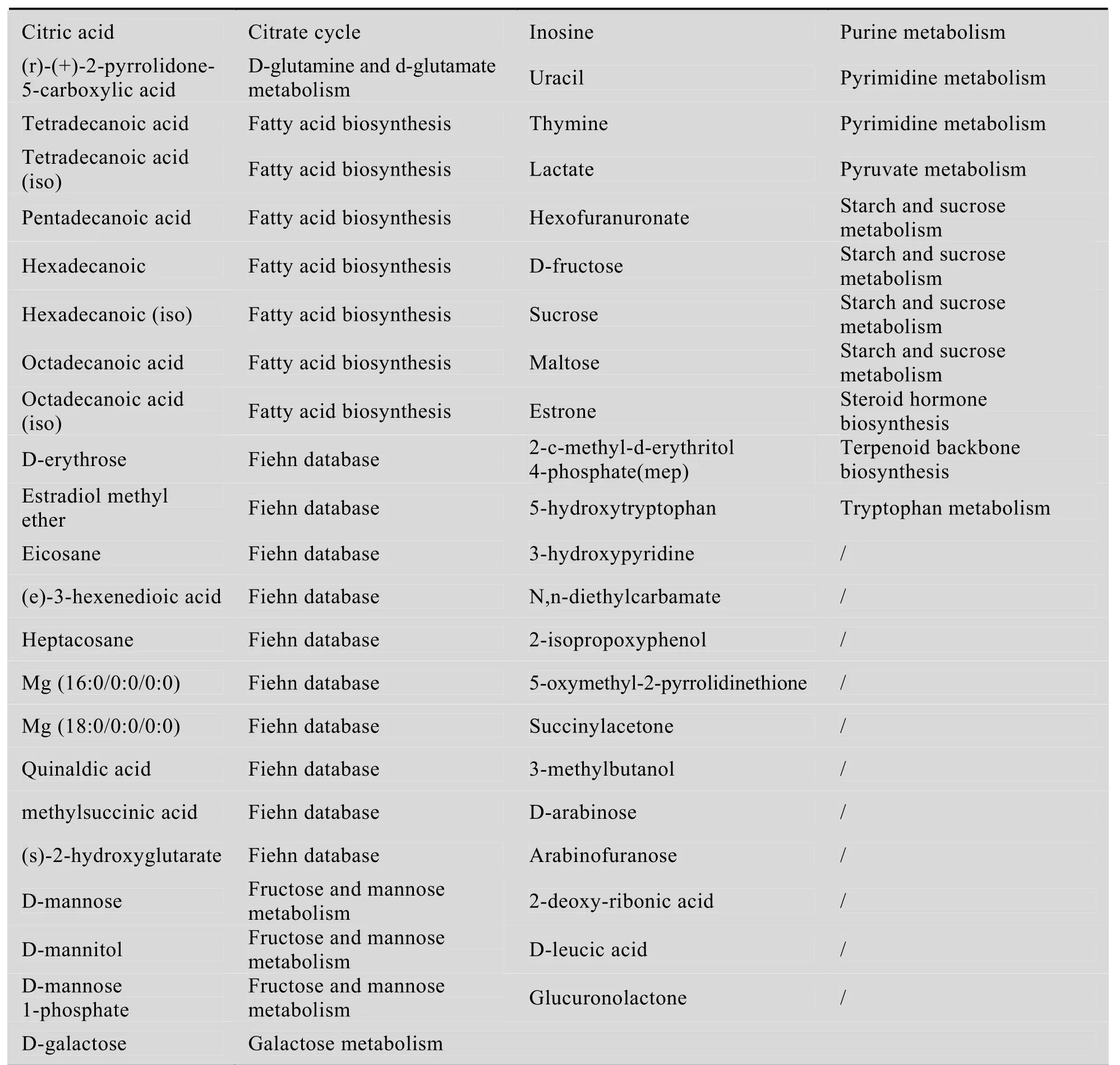
续表2

本工作对利用上述优化条件制备的代谢物样品进行了 GC-MS检测,结果如表 2所示。在能够检测到的400多个组分峰中,通过与 NIST、METLIN、Fiehn代谢物谱库以及分析,能够精确匹配糖酵解、戊糖磷酸途径、柠檬酸循环、氨基酸、脂肪酸及脂类、肌醇代谢、核酸代谢、糖苷类代谢、辅因子、甾醇、激素和萜类等代谢途径中的103个代谢物。
在大肠杆菌、枯草芽胞杆菌和酵母菌的代谢物组学研究中,一般能够检测到覆盖以上代谢途径的80–100个代谢物即能满足比较代谢物组学分析的要求[6-20]。因此我们认为利用我们建立的研究方法以及现有的质谱平台,也能够满足链霉菌代谢物组学相关研究。
2.4 天蓝色链霉菌细胞代谢的时序分析
建立了链霉菌代谢物组学研究方法后,为了解析链霉菌初级代谢和次级代谢的关联,我们对天蓝色链霉菌不同生长阶段部分代谢途径的相对强度进行了分析。首先,在R2YE液体培养基中测定了天蓝色链霉菌的生长曲线和次级代谢产物十一烷基灵菌红素和放线紫红素的产生曲线 (图 5)。可以看出,当菌体生长进入稳定期后,次级代谢产物十一烷基灵菌红素才开始产生 (约48 h),而放线紫红素的产生时间更滞后 (约72 h)。于是,根据前面建立的分析方法,选取对数前期 (18 h) 、对数期 (30 h)、稳定前期 (48 h) 和稳定后期 (72 h) 4个时间点分析该菌株的代谢时序差异。结果如图6所示,可以发现糖酵解途径、磷酸戊糖途径 (PPP)和三羧酸循环 (TCA) 在对数期有相对较高的代谢强度,而在次级代谢生物合成的稳定期(72 h),这些途径急剧减弱,可检测到的代谢物相对浓度仅有对数期 (30 h) 的 1%–16%;而可检测到的一些氨基酸相对于糖酵解途径和 TCA循环下降趋势较缓;与以上途径的相对强度趋势显著不同,相对于对数期和稳定后期,一些脂肪酸不仅在稳定前期浓度相对较高,而且在稳定后期的下降也相对缓慢,72 h代谢物浓度仍可达到30 h的63%–77%。这些结果表明,在链霉菌中部分氨基酸途径和脂肪酸途径可能充当了初级代谢和次级代谢转换的桥梁。
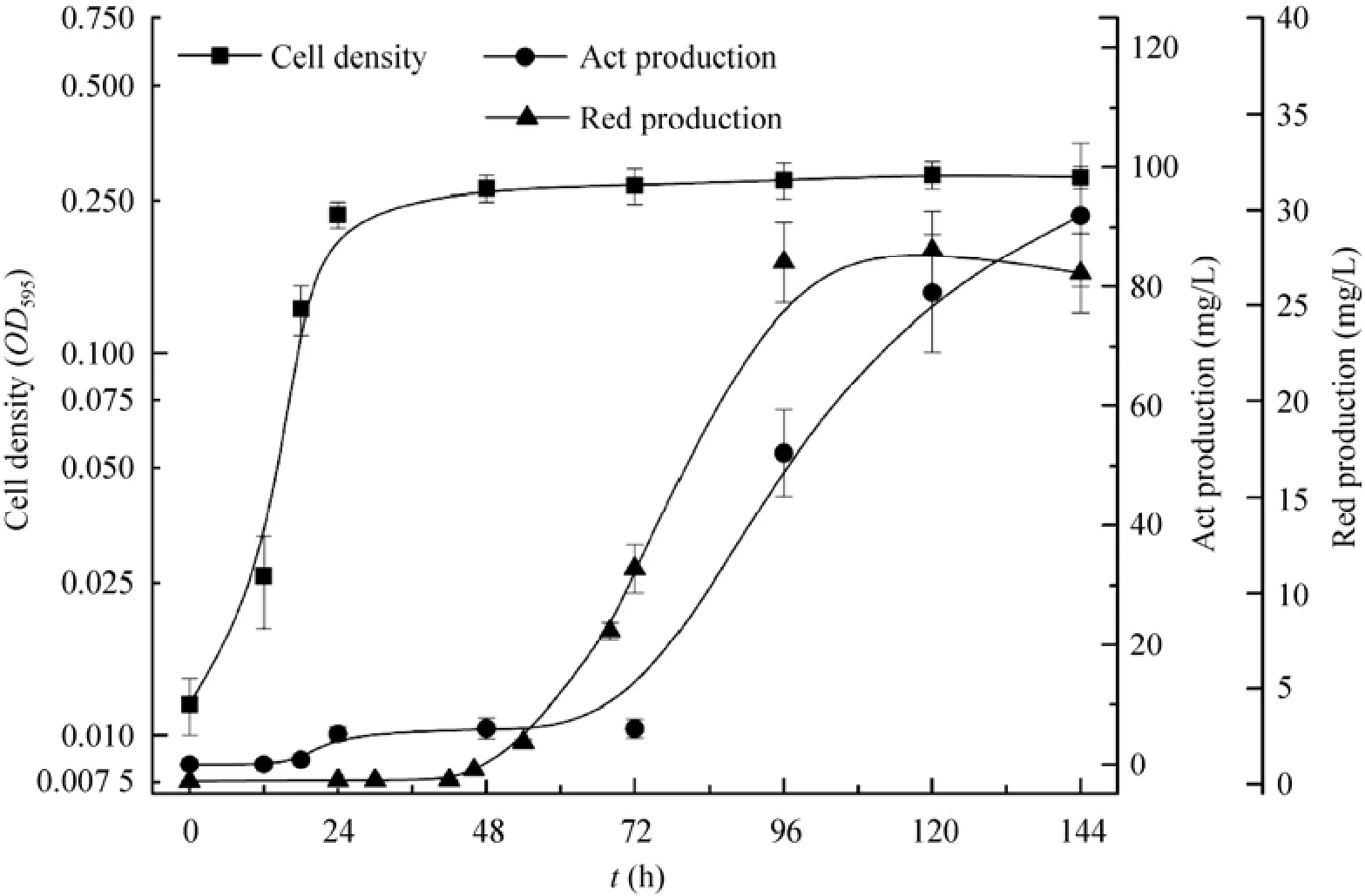
图5 天蓝色链霉菌M145生长曲线、放线紫红素和十一烷基灵菌红素合成曲线Fig. 5 Growth curve (■) , and production curves of undecylprodigiosin (▲), actinhordin (●) of S. coelicolor M145.
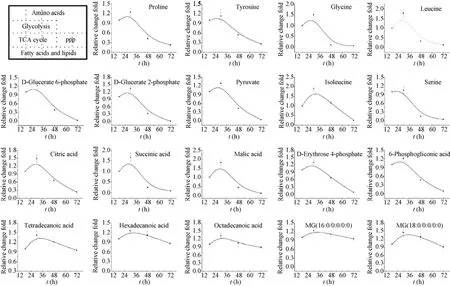
图6 天蓝色链霉菌M145主要胞内代谢物的时序变化特征Fig. 6 Temporal profiles of different metabolites in S. coelicolor M145.
3 讨论
链霉菌作为抗生素的重要工业产生菌,其代谢工程改造提高抗生素产量具有重要意义。在工程菌改造过程中,代谢物组学作为一种系统的研究手段,在目的产物相关代谢物的发现中具有无与伦比的优势。为了方便代谢物组学在链霉菌研究中的应用,本文对天蓝色链霉菌代谢物组学样品制备方法进行了一些摸索。由于链霉菌与其他细菌生长状态不同,会在液体培养基中形成菌丝团。这要求我们在制备准确反映细胞瞬时生理状态的代谢物时,不能采取和大肠杆菌、枯草芽胞杆菌等相同的方法,所以本文主要对链霉菌代谢物样品制备步骤进行了系统优化。
细胞淬灭能够及时终止胞内的代谢活动,是获得能够准确反映细胞瞬时生理状态的代谢物样品的关键环节。已有研究比较了各种淬灭溶剂对不同菌株代谢物提取的影响,并证明冷甲醇水对多种菌株具有较好的淬灭效果[13-21]。目前有关链霉菌代谢物组学的研究也多采用冷甲醇水为细胞淬灭剂[22-23]。由于淬灭剂会对细胞壁产生严重的破坏作用,因此严格控制淬灭时间防止导致胞内代谢物渗漏非常重要。然而目前并无相关研究考察以冷甲醇水为淬灭溶剂时,淬灭时间对链霉菌代谢物提取的影响。本研究发现,冷甲醇水的低温 (–40 ℃) 淬灭时间维持在 4 min时能够较好地保证及时终止链霉菌胞内的代谢活动,获得较多的代谢物。淬灭时间较短不能充分终止反应,而时间较长时,由于细胞壁受损严重导致胞内代谢物大量渗漏,致使获得的代谢物数目显著降低 (图 2)。此外,通过比较不同的菌体分离方法,发现快速抽滤法提取得到的代谢物质量优于离心分离法,产生这种结果的原因可能有以下两点:一,在离心过程中,细胞与淬灭溶剂接触时间较长,胞内代谢物与溶剂充分交换,导致大量渗漏;二,淬灭后的细胞壁非常脆弱,离心和洗涤过程产生的剪切力引发菌丝断裂,造成代谢物渗漏。相对而言,快速过滤时间短暂,可以避免胞内代谢物向溶剂大量扩散,此外该方法还可以避免剪切力对细胞损伤,减少代谢物渗漏。液氮反复冻融是提取胞内代谢物的主要方法[23-24],然而,对于具有较厚细胞壁的革兰氏阳性菌及真菌,反复冻融法通常不能充分提取胞内代谢物[8]。为了解决这一问题,我们采取了液氮研磨加反复冻融的提取方法。此外,研究还发现反复冻融的过程中较长的液氮速冻时间反而不利于代谢物的获得,这一现象的原因仍不确定。最后研究了温度对代谢物衍生率的影响,发现40 ℃条件下衍生化效率较37 ℃高。通过对各样品制备环节进行比较优化,最终得到最适合天蓝色链霉菌代谢物分析样品的制备方法。
通过我们建立的方法成功得到 400多个天蓝色链霉菌代谢物组分后,利用已有的NIST、MTEALIN和Fiehn代谢物数据库,参考相关文献并与KEGG数据库中该菌株的代谢途径相匹配,确定出 103个能与代谢途径精确匹配的代谢物。目前并无针对链霉菌胞内代谢物样品制备方法的详细研究,而利用现有的文献方法制备的代谢物样品,组分也主要集中于氨基酸及其衍生物和部分脂肪酸及脂类[23-25],少于本文方法所获得的组分种类。此外,其他未确定组分主要可分为两部分,一部分是与谱库中匹配度较低,只根据代谢物的精确质量数并不能定性;另一部分是天蓝色链霉菌的代谢途径数据库中并未收集该化合物信息。链霉菌不同于其他细菌,除了初级代谢,还存在大量的次级代谢基因簇,例如天蓝色链霉菌有23个次级代谢基因簇,这些基因簇的编码产物以及中间代谢物上没有相关的精确质谱信息,因此在链霉菌代谢物组学研究中,代谢物的精确匹配、归属仍存在较大挑战。
利用建立的链霉菌代谢物组学样品制备方法,我们对天蓝色链霉菌各个代谢途径在不同生长阶段的相对强度进行了分析,在代谢物层面上发现了链霉菌初级代谢相关途径 (糖酵解途径、戊糖磷酸途径和TCA循环) 和次级代谢的生物合成存在明显的时间差。而介于此时间差之间的是一些氨基酸和脂类物质的代谢物处于较高水平,所以我们推测可能是一些氨基酸和脂类相关途径在链霉菌生长周期中承担了衔接初级代谢和次级代谢的角色。
[1] Bentley SD, Chater KF, Cerdeno-Tarraga AM, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2).Nature, 2002, 417(6885): 141–147.
[2] Kell DB. Metabolomics and systems biology:making sense of the soup. Curr Opin Microbiol,2004, 7(3): 296–307.
[3] Wang QZ, Wu CY, Chen T, et al. Integrating metabolomics into a systems biology framework to exploit metabolic complexity: strategies and applications in microorganisms. Appl Microbiol Biotech, 2006, 70(2): 151–161.
[4] Dietmair S, Hodson MP, Quek LE, et al. Metabolite profiling of CHO cells with different growth characteristics. Biotech Bioeng, 2012, 109(6):1404–1414.
[5] Trushina E, Dutta T, Persson XMT, et al.Identification of altered metabolic pathways in plasma and csf in mild cognitive impairment and alzheimer’s disease using metabolomics. PLoS ONE, 2013, 8(5): e63644.
[6] Yuan J, Doucette CD, Fowler WU, et al.Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol,2009, 5: 302.
[7] Xia ML, Huang D, Li SS, et al. Enhanced FK506 production in Streptomyces tsukubaensis by rational feeding strategies based on comparative metabolic profiling analysis. Biotech Bioeng, 2013,DOI:10.1002/bit.24941.
[8] Kim S, Lee do Y, Wohlgemuth G, et al. Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae.Anal Chem, 2013, 85(4): 2169–2176.
[9] Bergdahl B, Heer D, Sauer U, et al. Dynamic metabolomics differentiates between carbon and energy starvation in recombinant Saccharomyces cerevisiae fermenting xylose. Biotechnol Biofuels,2012, 5(1):34.
[10] Cheng JS, Liang YQ, Ding MZ, et al. Metabolic analysis reveals the amino acid responses of Streptomyces lydicus to pitching ratios during improving streptolydigin production. Appl Microbiol Biotechnol, 2013, 97(13): 5943–5954.
[11] Sidebottom AM, Johnson AR, Karty JA, et al.Integrated metabolomics approach facilitates discovery of an unpredicted natural product suite from Streptomyces coelicolor M145. ACS Chem Biol, 2013, 8(9): 2009–2016.
[12] Li J, Ren LJ, Sun GN, et al. Gas chromatography-mass spectrometry (GC-MS) and its application in metabonomics. Chin J Biotech,2013, 29(4): 434–446 (in Chinese).李娟, 任路静, 孙冠男, 等. 气相色谱-质谱联用技术及其在代谢组学中的应用. 生物工程学报,2013, 29(4): 434–446.
[13] Winder CL, Dunn WB, Schuler S, et al. Global metabolic profiling of Escherichia coli cultures: an evaluation of methods for quenching and extraction of intracellular metabolites. Anal Chem, 2008,80(8): 2939–2948.
[14] Bolten CJ, Kiefer P, Letisse F, et al. Sampling for metabolome analysis of microorganisms. Anal Chem, 2007, 79(10): 3843–3849.
[15] Krall L, Huege J, Catchpole G, et al. Assessment of sampling strategies for gas chromatography-mass spectrometry (GC-MS) based metabolomics of cyanobacteria. J Chromatogr B, 2009, 877(27):2952–2960.
[16] Maud MK, Bas M, Mariet J, et al. Microbial metabolomics with gas chromatography-mass spectrometry. Anal Chem, 2006, 78(4):1272–1281.
[17] Zhao Y, Xiang S, Dai X, et al. A simplified diphenylamine colorimetric method for growth quantification. Appl Microbiol Biotech, 2013,97(11): 5069–5077.
[18] Ryu YG, Butler MJ, Chater KF, et al. Engineering of primary carbohydrate metabolism for increased production of actinorhodin in Streptomyces coelicolor. Appl Environ Microbiol, 2006, 72(11):7132–7139.
[19] Strauch E, Takano E, Baylts HA, et al. The stringent response in Streptomyces coelicolor A3(2). Mol Microbiol, 1991, 5(2): 289–298.
[20] Amador-Noguez D, Brasg IA, Feng XJ, et al.Metabolome remodeling during the acidogenic-solventogenic transition in Clostridium acetobutylicum. Appl Environ Microbiol, 77(22):7984–7997.
[21] Villas-Boas SG, Hojer-Pedersen J, Akesson M, et al. Global metabolite analysis of yeast: evaluation of sample preparation methods. Yeast, 2005,22(14): 1155–1169.
[22] Jankevics A, Merlo ME, de Vries M, et al.Metabolomic analysis of a synthetic metabolic switch in Streptomyces coelicolor A3(2).Proteomics, 2011, 11(24): 4622–4631.
[23] Kol S, Merlo ME, Scheltema RA, et al.Metabolomic characterization of the salt stress response in Streptomyces coelicolor. Appl Environ Microbiol, 2010, 76(8): 2574–2581.
[24] Wentzel A, Sletta H, Consortium S, et al.Intracellular metabolite pool changes in response to nutrient depletion induced metabolic switching in Streptomyces coelicolor. Metabolites, 2012, 2(1):178–194.
[25] Li Y, Florova G, Reynolds KA. Alteration of the fatty acid profile of Streptomyces coelicolor by replacement of the initiation enzyme 3-ketoacyl acyl carrier protein synthase III (FabH). J Bacteriol,2005, 187(11): 3795–3799.
猜你喜欢
现代临床医学(2022年4期)2022-09-29
传染病信息(2021年6期)2021-02-12
长江文艺(2020年9期)2020-10-23
少先队活动(2020年6期)2020-07-27
理化检验-化学分册(2020年5期)2020-06-15
农药科学与管理(2019年5期)2019-08-13
小学时代(2019年6期)2019-01-11
中成药(2017年12期)2018-01-19
安徽医科大学学报(2015年9期)2015-12-16
质谱学报(2015年5期)2015-03-01
