
治疗黑色素瘤新药
——Tafinlar
2014-03-07 11:24王丽君
药学研究 2014年1期
王丽君,王 真
(山东宏济堂制药集团有限公司,山东济南250010)
·药物信息·
治疗黑色素瘤新药
——Tafinlar
王丽君,王 真
(山东宏济堂制药集团有限公司,山东济南250010)
英国公司葛兰素史克(GSK)2013年5月29日宣布,黑色素瘤新药Tafinlar(dabrafenib)获得了FDA的批准。TAFINLAR为BRAF抑制剂,作为一种单药口服胶囊,适用于携带BRAF V600E突变的手术不可切除性黑色素瘤或转移性黑色素瘤成人患者的治疗。
英文名:Dabrafenib
中文名:达拉菲尼
商品名:Tafinlar
英文化学名:N-[3-[5-(2-Amino-4-pyrimidinyl)-2-(tert-butyl)-4-thiazolyl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide
中文化学名:N-[3-[5-(2-氨基-4-嘧啶基)-2-(叔丁基)-4-噻唑基]-2-氟苯基]-2,6-二氟苯磺酰胺
分子式:C23H20F3N5O2S2
分子结构:

1 作用机理
达拉菲尼为环吡酮胺(BRAF)激酶部分突变株的抑制剂,对BRAF V600E、BRAF V600K和BRAF V600D酶的体外半抑制浓度(IC50)分别为0.65,0.5和1.84 nM。达拉菲尼也是野生型BRAF和CRAF激酶抑制剂,对应的IC50分别为3.2和5.0 nM,对SIK1、NEK11和LIMK1激酶等亦有较高的抑制浓度。一些BRAF基因变异(包括导致BRAF V600E酶出现的基因变异)可导致结构性激活可激发肿瘤细胞生长的BRAF激酶。达拉菲尼在体内和体外均具有抑制BRAF V600突变阳性的黑色素瘤细胞生长。
2 药代动力学
2.1 吸收 口服用药后,中位达峰时间(Tmax)为2 h。口服达拉菲尼的平均绝对生物利用度为95%。单剂量用药12~300 mg范围内,达拉菲尼暴露量(血药峰浓度,Cmax和血药曲线下面积,AUC)具有剂量依赖性,但是低于同剂量每日两次用药,剂量依赖性的达拉菲尼暴露量。达拉菲尼150 mg每日两次重复用药后,平均蓄积率为0.73,稳态下,个体间的AUC变异率为38%。与快代谢人群相比,肥胖人群服用达拉菲尼后,Cmax和AUC分别下降51%和31%,平均Tmax延后3.6 h。
2.2 分布 达拉菲尼血浆蛋白结合率为99.7%,表观分布容积为70.2 L。
2.3 代谢 达拉菲尼主要通过肝药酶CYP2C8和CYP3A4转化为羟化达拉菲尼,羟化达拉菲尼可进一步被CYP3A4氧化为羧基达拉菲尼,进而通过胆汁和尿液排出体外。羧基达拉菲尼脱羰基形成去甲基达拉菲尼;去甲基达拉菲尼可在肠道被重吸收。去甲基达拉菲尼进一步被CYP3A4肝药酶代谢为氧化产物。羟化达拉菲尼和达拉菲尼的终末半衰期均为10 h,代谢产物羧基达拉菲尼和去甲基达拉菲尼的半衰期为21~22 h。重复给药时,平均代谢产物-母药AUC比值在羟化-,羧基-,去甲基-分别为0.9、11和0.7。基于系统暴露、相对效价和药动学特点,可以认为羟化达拉菲尼和去甲基达拉菲尼是达拉菲尼发挥临床治疗作用的活性物质。
2.4 消除 口服达拉菲尼的终末半衰期为8 h。达拉菲尼单剂量用药的表观清除率为17.0 L·h-1,每天2次用药,连续给予2周达拉菲尼,这时的表观清除率为34.4 L·h-1。根据放射同位素得到的数据显示,所有代谢产物由71%通过粪便排出,23%通过尿液排出体外。
2.5 药物相互作用 达拉菲尼为肝药酶CYP3A4和CYP2C8的作用底物,羟化达拉菲尼和羧基达拉菲尼为CYP3A4的作用底物。体外研究中,达拉菲尼是人P-糖蛋白(P-glycoprotein,Pgp)和乳腺癌对偶蛋白(breast cancer resistance protein,BCRP)的作用底物。在人体肝细胞里,达拉菲尼可剂量依赖性诱导CYP2B6和CYP3A4 mRNA表达增加(可达对照组的32倍),体内对CYP3A4具有中度诱导作用。体外研究中,达拉菲尼及其代谢产物(羟化-,羧基
-,去甲基-)为有机阴离子转运多肽OATP1B1,OATP1B3,
OAT1和OAT3的抑制剂;同时为BCRP的中度抑制剂。
3 动物毒理学与药理学
3.1 致癌、致畸和致突变效应 达拉菲尼未进行致癌性研究。临床研究显示,达拉菲尼能增加皮肤鳞状细胞癌变的风险。体外细菌回复试验(Ames test)和小鼠淋巴瘤测定显示,达拉菲尼无致突变效应。大鼠骨髓细胞微核试验显示达拉菲尼为致畸效应。
雌性小鼠生殖能力与胚胎存活数研究中,给予20 mg·kg-1·d-1(根据AUC计算,此剂量相当于临床推荐剂量)或以上剂量时,小鼠生殖能力降低;给予300 mg·kg-1·d-1(根据AUC计算,相当于临床推荐剂量时的3倍),小鼠生殖能力降低;怀孕雌性小鼠卵黄体数减少。
达拉菲尼对雄性生育能力影响的研究未进行,但是,在连续用药研究中,分别给予相当于临床推荐剂量或三倍临床推荐剂量情况下,达拉菲尼可有使大鼠或犬双侧睾丸退化或衰竭的作用。
3.2 临床前毒理学/药理学研究 给予50 mg·kg-1·d-1(根据AUC计算,大约相当于临床推荐剂量时的5倍)或以上剂量,用药至4周时,犬出现心血管不良反应。这类不良反应包括冠脉退变/梗死和出血,心脏房室瓣膜肥大/出血。
4 临床研究
一项多国家、多中心、随机(3∶1)、开放、阳性对照试验,对达拉菲尼的安全性和有效性进行了评估,本试验共纳入250例未经过前期治疗BRAF V600E突变阳性、未切除或转移性黑色素瘤患者。任何经过BRAF抑制剂或MEK抑制剂治疗的患者排除在外。患者随机分为两组:达拉菲尼组(n=187)每日150 mg,分两次口服;达卡巴嗪组(n=63)每3周一次,按1 000 mg·m-2静脉给药。所有患者按肿瘤非切除状态III级[局部结节或正在发生转移,M1a:远处皮肤、皮下或转移性结节,或M1b:肺转移vs M1c:黑色素瘤(所有其他出现内脏转移或血清LDH升高者)]为基线进行分层。本研究的主要评价终点为经研究人员确认的无进展生存率(PFs)。另外,一个独立的放射学审查委员会(IRRC)通过预先确认的支持性分析结果如PFS,确认的客观反应率(ORR)及反应期等进行治疗的有效性评估。
纳入试验的患者中位年龄为52岁,主要为男性(60%)、白人(99%)、ECOG评分0分(67%)、M1c期(66%)、LDH正常(62%)。所有患者的BRAF V600E变异肿瘤组织均在集中的试验点通过临床分析确认。243例患者(97%)的肿瘤组织采用FDA认可的诊断指南(THxID-BRAF分析)进行回顾性确认见表1,图1。
5 规格与剂量
Tafinlar为胶囊剂型,口服用药,上市有50 mg和75 mg两个规格。每50 mg制剂中含有59.25 mg的硫酸达拉菲尼(相当于50 mg的达拉菲尼),每75 mg制剂中含有88.88 mg的硫酸达拉菲尼(相当于75 mg的达拉菲尼)。
剂量和给药方法:①开始用Tafinlar治疗前确证在肿瘤标本中存在BRAFV600E突变;②推荐剂量150 mg·d-1,分2次口服,餐前至少1 h或餐后至少2 h服用。
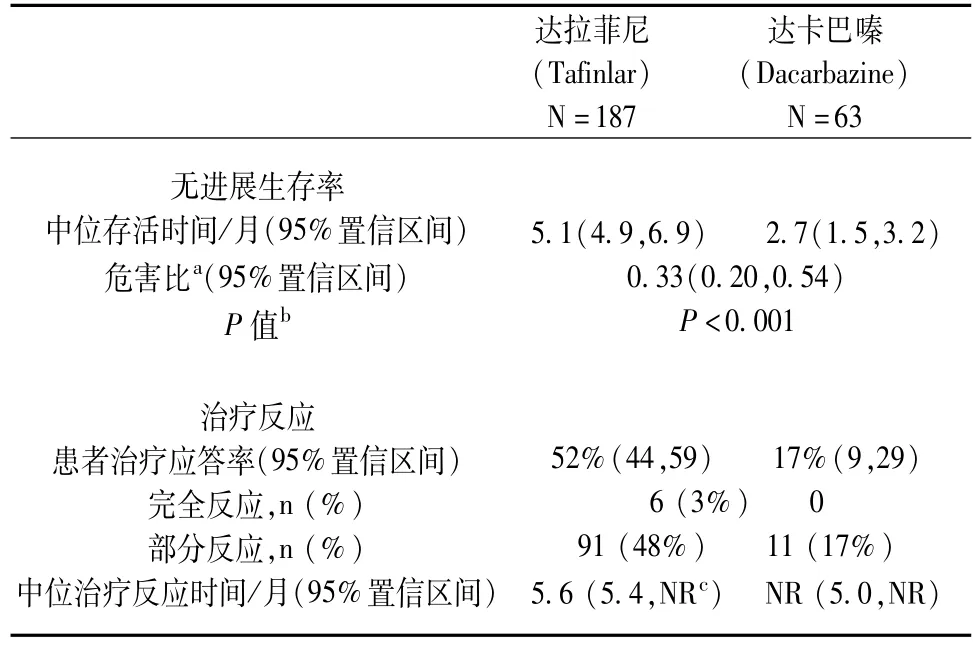
表1 研究人员确认的无进展生存率(基于CTA)

图1 研究人员确认的无进展生存率Kaplan-Meier曲线(基于CTA的意向性治疗人群)
6 适应证
Tafinlar是一种激酶抑制剂,适用于有不能切除或转移的BRAF V600E突变(FDA批准监测方法确认)的黑色素瘤患者的治疗。
使用限制:Tafinlar不适用于有野生型BRAF黑色素瘤患者的治疗。
7 不良反应
对Tafinlar最常见不良反应(≥20%)是角化过度、头痛、发热、关节炎、乳头状瘤、脱发和掌跖红肿疼痛综合征(PPES)。
8 警告和注意事项
①新原发性皮肤恶性病:开始治疗前、治疗时每3个月和停止Tafinlar后直至6个月进行皮肤学评价;②在BRAF野生型黑色素瘤中促肿瘤:用BRAF抑制剂可能发生细胞增殖增加;③严重发热性药物反应:出现发热≥101.3°F(38.5℃)或发生并发发热立即停用Tafinlar;④高血糖:糖尿病或高血糖患者应监测血糖水平;⑤葡萄膜炎和虹膜炎:常规监测患者视力症状;⑥葡萄糖-6磷酸脱氢酶缺乏:严密监测溶血性贫血;⑦胚胎胎儿毒性:可能致胎儿危害。
R979.1
A
2095-5375(2014)01-0061-002
王丽君,女,研究方向:药品检验,E-mail:281960430@qq.com
猜你喜欢
昆明医科大学学报(2022年8期)2022-07-31
意林(2022年2期)2022-02-13
实用肿瘤学杂志(2020年6期)2020-12-09
世界知识画报·艺术视界(2020年6期)2020-07-04
植物营养与肥料学报(2019年12期)2019-03-07
西南国防医药(2016年6期)2016-12-01
橡胶工业(2016年2期)2016-02-23
青岛画报(2015年8期)2015-09-17
中国医学创新(2015年33期)2015-04-27
中国药理学通报(2014年2期)2014-05-09
