
Improved preparation and chemical kinetics on fully automated synthesis of[18F]-THK523,a PET imaging probe for Tau pathologies∗
2014-03-07 12:24:23KONGYanYan孔艳艳SIZhan司展CAOGuoXian曹国宪ZHANGZhengWei张政伟WUPing吴平XUEFangPing薛方平DUFuQiang杜富强ZHUJianHua朱建华LICong李聪CHENJian陈键andGUANYiHui管一晖
Nuclear Science and Techniques 2014年4期
KONG Yan-Yan(孔艳艳),SI Zhan(司展),CAO Guo-Xian(曹国宪), ZHANG Zheng-Wei(张政伟),WU Ping(吴平),XUE Fang-Ping(薛方平),DU Fu-Qiang(杜富强), ZHU Jian-Hua(朱建华),LI Cong(李聪),CHEN Jian(陈键),and GUAN Yi-Hui(管一晖),
1PET Center,Huashan Hospital,Fudan University,Shanghai 200235,China
2Key Laboratory of Nuclear Medicine,Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine,Wuxi 214063,China
3Key Laboratory of Smart Drug Delivery,Ministry of Educationamp;PLA, School of Pharmacy,Fudan University,Shanghai 200032,China
Improved preparation and chemical kinetics on fully automated synthesis of[18F]-THK523,a PET imaging probe for Tau pathologies∗
KONG Yan-Yan(孔艳艳),1SI Zhan(司展),1CAO Guo-Xian(曹国宪),2ZHANG Zheng-Wei(张政伟),1WU Ping(吴平),1XUE Fang-Ping(薛方平),1DU Fu-Qiang(杜富强),1ZHU Jian-Hua(朱建华),3LI Cong(李聪),3CHEN Jian(陈键),3and GUAN Yi-Hui(管一晖)1,†
1PET Center,Huashan Hospital,Fudan University,Shanghai 200235,China
2Key Laboratory of Nuclear Medicine,Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine,Wuxi 214063,China
3Key Laboratory of Smart Drug Delivery,Ministry of Educationamp;PLA, School of Pharmacy,Fudan University,Shanghai 200032,China
Extensive accumulation of neurofibrillary tangles(NFTs)consistently correlate with the degree of cognitive impairment and neuronal circuitry deterioration associated with Alzheimer’s disease.However,no PET probe is currently available for selective detection of NFTs in the living human brain.[18F]-THK523 was developed as a potential in vivo imaging probe for tau pathology.In this paper,we report a new protected precursor,2-((2-(4-((tert-butoxycarbonyl)amino)phenyl)quinolin-6-yl)oxy)ethyl 4-methylbenzenesulfonate(THK-7),instead of 2-((2-(4-aminophenyl)quinolin-6-yl)oxy)ethyl 4-methylbenzenesulfonate(BF241),and an improved automated radiosynthesis of[18F]-THK523 and the study on chemical kinetics of the labeling reaction of[18F]-THK523, with high-yield(70±5%,n=6,decay-corrected to end of bombardment),and high radiochemical purity (>90%)and specific activity(2.5±0.5Ci/µmol)from protected precursor on fully automated module at the end of radiosynthesis(45–55min).The chemical kinetics for[18F]-THK523 demonstrates that nucleophilic substitution can be carried out easily with protected precursor.
Fluorine-18 radiolabeling,Neurofibrillary tangles,Alzheimer’s disease,Automated radiosynthesis,Chemical kinetics
I.INTRODUCTION
Alzheimer’s disease(AD)is characterized by progressive impairment of cognitive abilities,such as memory,learning, and social skills.The effects of AD are devastating for both the patients and their family,affecting their everyday life.AD mostly afflicts the elderly,resulting in an economic challenge for our healthcare system,as the elderly population grows[1].
A clinical diagnosis of AD is assessed by several tools, such as medical history and neuropsychological criteria(e.g. National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association)[2].However,a definitive diagnosis of AD can only be made by confirming the presence of cerebral extracellular senile plaques(SPs)and intracellular neurofibrillary tangles(NFTs)from a postmortem assessment.SPs and NFTs that consist of amyloid-β peptides (Aβ)and paired helical filaments(PHFs)of hyperphosphorylated tau protein are neuropathological hallmarks in AD. Tau protein is a microtubule-associated protein present in ax-ons,the roles of which include stabilization of the microtubules,modulation of the plasticity of the cytoskeleton,and the promotion of neurite outgrowths.When hyperphosphorylated,tau proteins aggregate into PHFs and NFTs,resulting in the destabilization of microtubules and disruption of axonal transport[3–5].Eventually,these stresses will cause neuronal deterioration and ultimately neuronal death.The severity of NFTs accumulation correlates with the degree of cognitive impairment and neuronal deterioration associated with AD[6–9].This correlation makes NFTs potential biomarkers that can be targeted to study how the AD pathology progresses and its association with cognitive deterioration[3,10,11].
The detection of NFTs in the early stages of AD might be of great value for diagnostic and treatment purposes. Positron emission tomography(PET)is a noninvasive diagnostic imaging modality,which utilizes radioisotopelabeled target molecular probes and is considered as a diagnostic tool that enables early detection of pathologies. Synthesizing molecular probes with a high specificity and affinity to NFTs for PET imaging can allow an earlier diagnosis and monitoring the progression of AD in vivo[12]. Reports on PET imaging agents selectively targeting NFTs include:radioiodinated styrylbenzimidazole(SBIM)derivative[13];radioiodinated phenyldiazenylbenzothiazole(PDB) derivative[14];quinoline and benzimidazole derivatives, [11C]BF158[15]and[18F]-THK523[16];rhodanine and thiohydantoin derivative[17];phenyldiazenyl benzothiazole deriveatives,[18F]T807/808[3,18]and[18F]-(E)-4-((6-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)benzo[d]thiazol-2-yl)-diazenyl)-N,N-dimethylaniline([18F]FPPDB)[19],and 2-(1- 6-[(2-[F]fluoroethyl)(methyl)amino]-2-naphthylg ethylidene)malononitrile [18F]FDDNPg[20].One of the imaging agents,[18F]-THK523,displayed high affinity and selectivity for tau pathology both in vitro and in vivo[16,21].In this paper,we elevate the automated radiosynthesis and reaction kinetics of[18F]-THK523 from a new precursor,2-((2-(4-((tert-butoxycarbonyl)amino)phenyl)quinolin-6-yl)oxy)ethyl 4-methylbenzenesulfonate(THK-7).This differs from the previous synthesis of[18F]-THK523 by Fodero-Tavoletti MT(Fig.1),which utilized 2-((2-(4-aminophenyl)quinolin-6-yl)oxy)ethyl4-methylbenzenesulfonate (BF241)asa precursor[17].

Fig.1.Structure of precursor.
II.EXPERIMENTAL
A.Reagents and instrumentation
KryptofixTM2.2.2,sodium bicarbonate,sodium hydroxide and hydrochloric acid of analytical grade were purchased from Sigma-Aldrich Corporation(St.Louis,MO,USA). Acetonitrile and ethanol of HPLC grade were obtained from Shanghai Lingfeng Chemical Reagent Co.,Ltd.(Shanghai, China).Sep-Pak tC18 solid phase extraction(SPE)cartridge (78.4µm of particle size)and sterile filters(0.22µm)were purchased from Waters Corporation(Milford,Massachusetts, USA).
Semi-preparative high-performance liquid chromatography was conducted using a Waters pump(Waters Corporation,Milford,Massachusetts,USA)with a Bioscan radioactivity detector.Analytical radio-HPLC(Waters Corporation) was equipped with a dual λ absorbance detector(Waters 2487),binary HPLC pump(Waters 2487)and a Bioscan radioactivity detector.TLC plate radioactivity was measured on a Wizard 1470 automatic gamma counter(U.S.Perkin Elmer Company)equipped with a multi-channel analyzer. The [18F]-THK523 synthesis module(PET-IT-I Reactor Module) was purchased from PET Scienceamp;Technology Co.Ltd. (Beijing,China).NMR and LC-MS were purchased from Bruker Corporation(Germany).
B.Precursor synthesis
1. Preparation of 6-methoxy-2-(4-nitrophenyl)quinolone:THK-1
A mixture of 4-nitro-cinnamic aldehyde (20g, 112.9mmol)and methoxyphenethylamine(25g,203mmol) were mixed into 37%HCl(70mL)under nitrogen,and the mixture was heated to 140◦C reflux for 3h.The solution was poured into ice water and adjusted to pH 8 with ammonia. The aqueous layer was extracted with three portions of ethyl acetate.The combined organic layer was washed with saturated sodium chloride,dried,filtered and concentrated. The residue was purified by flash column chromatography (N-hexane:Dichloromethane=1∶1)to afford THK-1 (8.8g,31.4mmol)in 28%yield.1H NMR(300MHz, CDCl3)δ 8.30–8.40(m,4H),8.18(d,1H,J=9.3Hz),8.08 (d,1H,J=9.3Hz),7.89(d,1H,J=8.6Hz),7.43(dd, 1H,J=8.3,2.8Hz),7.12(d,1H,J=2.8Hz);LC-MS: calculated for C16H12N2O3,280.08;found[M+H]281.0.
2. Preparation of 2-(4-nitrophenyl)quinolin-6-ol:THK-2
THK-1(3g,10.7mmol)was added to 30%HBr(260mL) and heated to 125–130◦C reflux for 3h. The solution was alkalized by NaHCO3and extracted with ethyl acetate.The combined organic layer was washed with saturated sodium chloride,dried,filtered and concentrated. The residue was purified by flash column chromatography (Dichloromethane∶Ethyl acetate=10∶1)to afford THK-2 (2.35g,8.8mmol)in 82.5%yield.1H NMR(300MHz,d-DMSO)δ 10.2(s,1H),8.51(d,2H,J=9.2Hz),8.37(d, 2H,J=9.0Hz),8.33(d,1H,J=8.5Hz),8.16(d,1H, J=8.6Hz),7.98(d,1H,J=9.1Hz),7.39(dd,1H,J=9.1, 2.7Hz),7.21(d,1H,J=2.7Hz);LC-MS:calculated for C16H12N2O3,266.07;found[M+H]267.0.
C.Preparation of 6-(2-((tert-butyldiphenylsilyl)oxy)ethoxy)-2-(4-nitrophenyl)quinoline: THK-3
THK-2(2.35g,8.8mmol)was dissolved in 80mL acetonitrile.To the solution were added K2CO3(14.6g,105.6mmol) and TBDPSOCH2CH2Br(4.8g,13.2mmol).The mixture was heated to 90◦C reflux for 16h under nitrogen.The solution was filtered,washed with dichloromethane,and evaporated. The residue was purified by flash column chromatography(Dichloromethane∶N-hexane=1∶1)to afford compound THK-3(4.1g,7.5mmol)in 84.7%yield.1H NMR(300MHz,CDCl3)δ 8.31–8.59(m,4H),8.14(d, 1H,J=8.4Hz),8.07(d,2H,J=8.3Hz),7.88(d,1H, J=8.5Hz),7.71–7.74(m,4H),7.35–7.45(m,7H),7.09 (d,1H,J=2.7Hz),4.25(t,2H,J=5.1Hz),4.09(t,2H, J=5.0Hz),1.08(s,9H).
D. Preparation of 4-(6-(2-((tertbutyldiphenylsilyl)oxy)ethoxy)quinolin-2-yl)aniline: THK-4
THK-3(3g,5.5mmol)was dissolved in 300mL ethanol and cooled to 0◦C with ice-bath. Anhydrous Cu(OAc)2(1.31g,7.2mmol)was added to the solution. NaBH4(10.41g,275.2mmol)was added portion wise within 25min. The mixture was cooled for 10min in ice-bath.Then the solution was warmed to room temperature and stirred for 3.5h. All the above reactions were carried out under nitrogen.The mixture was poured into 50mL water and ethanol was evaporated.The aqueous layer was extracted with ethyl acetate. The organic layer was washed,dried,filtered and concentrated to afford THK-4(2.4g,4.6mmol)in 83%yield.1H NMR(300MHz,CDCl3)δ 7.97–8.01(m,4H),7.71–7.76 (m,5H),7.30–7.43(m,7H),7.02(d,1H,J=2.8Hz),6.80 (d,2H,J=8.6Hz),4.21(t,2H,J=5.1Hz),4.07(t,2H, J=5.1Hz),3.80(brs,2H),1.07(s,9H);LC-MS:calculated for C33H34N2O2Si,518.24;found[M+H]519.2.
E.Preparation of tert-butyl(4-(6-(2-((tertbutyldiphenylsilyl)oxy)ethoxy)quinolin-2-yl)phenyl) carbamate:THK-5
THK-4(2.4g,4.6mmol),Boc2O(3g,13.7mmol)and triethylamine(1.8g,17.8mmol)were dissolved into tetrahydrofuran(THF)(30mL)and heated to 90◦C reflux for 16h under the protection of nitrogen.The solvent was removed under reduced pressure.The residue was purified by column chromatography(Dichloromethane∶Ethyl acetate=8∶14∶1)to afford THK-5(2.4g,3.9mmol)in 84%yield.1H NMR(300MHz,CDCl3)δ 8.09(d,2H,J=8.8Hz),8.03 (d,1H,J=8.6Hz),8.02(d,1H,J=9.1Hz),7.79(d,1H, J=8.6Hz),7.70–7.75(m,4H),7.51(d,2H,J=8.7Hz), 7.32–7.45(m,7H),7.04(d,1H,J=2.8Hz),6.59(s,1H), 4.22(t,2H,J=5.1Hz),4.07(t,2H,J=5.1Hz),1.55(s, 9H),1.07(s,9H).
F.Preparation of tert-butyl (4-(6-(2-hydroxyethoxy)quinolin-2-yl)phenyl)carbamate: THK-6
THK-5(1.01g,1.6mmol)was dissolved in 60mL THF. Bu4NF·3H2O(2g,6.3mmol)in 10mL THF was added to the solution.The reaction was allowed to stir at room temperature for 15–20min under nitrogen.The solvent was removed under reduced pressure.The residue was purified by column chromatography(Dichloromethane∶Ethyl acetate=4∶1) to afford THK-6(0.5g,1.3mmol)in 65%yield.1H NMR (300MHz,CDCl3)δ 9.55(s,1H),8.27(d,1H,J=8.5Hz), 8.13(d,2H,J=8.8Hz),8.02(d,1H,J=8.7Hz),7.93(d, 1H,J=8.9Hz),7.61(d,2H,J=8.8Hz),7.35–7.42(m, 2H),4.93(t,1H,J=5.5Hz),4.13(t,2H,J=4.8Hz),3.80 (dd,2H,J=4.8,5.4Hz),1.50(s,9H).
G. Preparation of 2-((2-(4-((tertbutoxycarbonyl)amino)phenyl)quinolin-6-yl)oxy)ethyl 4-methylbenzenesulfonate:THK-7
THK-6(0.48g,1.2mmol)was dissolved in 27mL dichloromethane.After the mixture was cooled to−5◦C in an ice/sodium chloride bath,triethylamine(0.842g, 8.3mmol)and DMAP(0.154g,1.3mmol)were added.TsCl (1.07g,5.6mmol)was added at−10◦C.Then the ice bath was removed and the mixture was allowed to stir overnight at room temperature.The above reaction was carried out under N2atmosphere.The solvent was removed under reduced pressure.The residue was purified by column chromatography(Dichloromethane∶Ethyl acetate=30∶1!8∶1) to afford 600mg of a yellow solid.Finally,pure product THK-7(0.59g,1.1mmol)in 87.5%yield was crystallized from Ethyl acetate(20mL)and N-hexane(35mL).1H NMR (300MHz,CDCl3)δ 9.56(s,1H),8.23(d,1H,J=8.7Hz), 8.14(d,2H,J=8.8Hz),8.03(d,1H,J=8.8Hz),7.91 (d,1H,J=9.0Hz),7.82(d,2H,J=8.3Hz),7.62(d,2H, J=8.8Hz),7.46(d,2H,J=8.0Hz),7.24–7.30(m,2H), 4.42–4.45(m,2H),4.30–4.33(m,2H),2.39(s,3H),1.50 (s,9H);LC-MS:calculated for C29H30N2O6S,534.18;found [M+H]535.0.
H.Standard synthesis for[18F]-THK523,tert-butyl (4-(6-(2-fluoroethoxy)quinolin-2-yl)phenyl)carbamate (THKF-2)
1. Preparation of 6-fluoro-2-(4-nitrophenyl)quinoline:THKF-1
THK-2(0.5g,1.9mmol),K2CO3(2.6g,18.8mmol)and BrCH2CH2F(0.5g,3.9mmol)were added to 24mL acetonitrile.The mixture was refluxed for 16h at room temperature under nitrogen.The solution was filtered and evaporated.The residue was purified by column chromatography (N-hexane∶Ethyl acetate=4∶1!3∶1)to afford THKF-1(0.55g,1.8mmol)in 93.4%yield.1H NMR(300MHz, CDCl3)δ 8.31–8.39(m,4H),8.18(d,1H,J=8.5Hz),8.12 (d,1H,J=9.3Hz),7.90(d,1H,J=8.6Hz),7.48(dd,2H, J=9.2,2.8Hz),7.14(d,2H,J=2.8Hz),4.86(dm,2H, J=47.4Hz),4.36(dm,2H,J=27.6Hz).

Fig.2.Schematic diagram for synthesis of[18F]-THK523.
2. Preparation of tert-butyl (4-(6-(2-fluoroethoxy)quinolin-2-yl)phenyl)carbamate:THKF-2
THKF-1(0.55g,1.8mmol)was dissolved in ethanol (120mL)under nitrogen.The solution was cooled to 0◦C and then anhydrous Cu(OAc)2(0.42g,2.3mol)was added. NaBH4(2g,52.9mol)was added portionwise at−5◦C.The solution was allowed to stir at−5◦C for 30min.Then the mixture was allowed to warm up to room temperature while stirring for 1h.The solvent was evaporated and the mixture was poured into 50mL water.The aqueous layer was extracted with ethyl acetate.The organic layer was washed, dried,filtered and concentrated.The residue was purified by flash column chromatography(N-hexane∶Ethyl acetate= 6∶1!3∶1)to afford THKF-2(0.21g,0.7mmol)in 42% yield.1H NMR(300MHz,CDCl3)δ 7.97–8.04(m,4H),7.76 (d,1H,J=8.7Hz),7.39(dd,1H,J=9.3,2.7Hz),7.07(d, 1H,J=2.7Hz),6.80(d,1H,J=8.6Hz),4.88(dm,2H, J=47.4Hz),4.34(dm,2H,J=27.6Hz);LC-MS:calculated for C17H15FN2O,283.13;found[M+H]283.0.
I.Radiosynthesis of[18F]-THK523
[18F]-THK523 was synthesized using an automated module(PET Scienceamp;Technology Co.Ltd.,Beijing China) (Fig.2).The no carrier added aqueous[18F]fluoride–was produced by the18O(p,n)18F nuclear reaction on an Eclipse HP Cyclotron(Siemens).
The synthesis of[18F]-THK523 includes five steps:1) azeotropic evaporation,2)nucleophilic substitution,3)hydrolysis for deprotection,4)purification,and 5)sterile filtration(Fig.3).Briefly,[18F]fluoride was trapped on a QMA cartridge and eluted into the reaction vial with 1.5mL mixture of K2CO3in water and KryptofixTM2.2.2 in Acetonitrile(19/31mmol/L)which was pre-added in bottle 1#.The mixture in the vial was evaporated at 116◦C for 206s under a N2flow and was co-evaporated to dryness with anhydrous acetonitrile(2mL)in bottle 2#at 116◦C for another 203s.The tosylated precursor(2mg in 1mL acetonitrile)in bottle 3#was added to the dried K[18F]and the nucleophilic substitution reaction was carried out at 120◦C for 15min to afford[18F]-THK.After excess of acetonitrile was removed at 120◦C under a flow of nitrogen,HCl(1mol/L,250µL)in bottle 4#was added to hydrolyze the Boc protecting group. The mixture was allowed to react at 105◦C for 5min,and NaOH(2mol/L,125µL)stored in bottle 5#was added followed by saturated NaHCO3(1mol/L,125µL)in bottle 6#to neutralize the solution.
The neutralized solution was loaded on a C-18 Sep-Pak,which was further washed with water to remove free18F–,KryptofixR?TM2.2.2,and other polar byproducts.The cartridge was then eluted with ethanol(2mL). The obtained crude product was collected and injected onto a semipreparative column(Waters XBridgeTMprep Shield RP18 10µm,250mm×10mm,part No.186003990,serial No. 101/123041GG01)at a flow rate of 4mL/min(70%EtOH: 30%H2O).The fraction containing[18F]-THK523 was collected from 6.0–6.5min on semi-preparative HPLC and was evaporated to dryness.To the residual,10%ethanol in saline (5mL)was added and the resulting solution was stabilized with ascorbic acid(2mg,0.011mmol)before sterile filtration through a 0.22µm membrane filter into a sterile vial.

Fig.3.Radiosynthesis of[18F]-THK523.
J.Quality control of[18F]-THK523
The labeling yield was determined by thin layer chromatography(TLC).Apply 2–4µL of the crude reaction solution to an activated silica gel G60 with?uorescence (F254)plates.Develop the chromatogram in a solvent system consisting of a mixture of ethyl acetate∶n-hexane∶triethylamine=4∶1∶0.005(v/v)until the solvent has moved about 3/4 of the length of the hromotogram.Remove the chromatogram,and allow the chromatogram to dry at room temperature.Determine the radioactivity distribution by cutting the chromatogram into 10 pieces of strips with equal length from sampling spot to where the solvent developed and counting each strip in a Wizard 1470 automatic gamma counter(U.S.Perkin Elmer Company)equipped with a multi-channel analyzer.The Rf value of[18F]-THK523 was 0.7–0.8,and Rf value of free F-18 was 0.0.Two TLCs were run for each tested reaction condition and radiolabelling yields were obtained by averaging the yields of the two runs.
Radiochemical purity(RCP)was determined by analytical radio-HPLC(high-performance liquid chromatography).The final product(20µL)was injected into the HPLC column(PurospherR?STAR LPRP-18e endcapped(5µm), 250mm×4.6mm,sorbent Lot No.TA1752311,column No. 210072,acetonitrile/0.05%triethylamine in water= 8/2 (v/v),flow rate at 0.6mL/min)at room temperature.The absorbance measured at 350nm retention time(tR)of the standard and[18F]-THK523 were 6.12 and 5.93min,respectively. The chemical identity was verified by co-injection with cold standard THKF-2.
K.Studies of reaction kinetics
The reaction conditions for the nucleophilic substitution were optimized by studying the reaction kinetics.Six vials, each containing a 30–50mCi activity of18F–,were used. After azeotropic evaporation,THK-7(1mL,3.7mmol/L)in DMSO/CAN(1/5)was added to the vials and reacted at 25◦C,120◦C and 160◦C according to the procedure described above. Three reaction times,2min,15min and 30min,were investigated at each temperature.After the same deprotecting reaction workup,solutions were sampled with a capillary and labeling yields were determined TLC.Films were dried and cut into 10 sections,then counted with γcounter.Order of reaction(n),rate constant(k)and activation energy(Ea)of the labeling reaction were calculated with the CHEMKIN code,developed by Cao et al.[22],to quantitatively study the labeling reaction for optimal conditions of nucleophilic substitution.
The rate constant,k,for the formation of[18F]-THK523, was calculated as follows.The reaction can be expressed as (let a=[K18F]0,b=[THK]0,x=[18F−THK523]t):

Integrating of each side of Eq.(3)between t=0!t and x=0!x gives:

Since a≈ x=10-9∼ 10-8mol/L,b=10-5∼10-4mol/L,b?a≈x,Eq.(4)can be simplified as


Fig.4.Synthesis of 2-((2-(4-((tert-butoxycarbonyl)amino)phenyl)quinolin-6-yl)oxy)ethyl 4-methylbenzenesulfonate(THK-7).

Fig.5.Synthesis of tert-butyl(4-(6-(2-fluoroethoxy)quinolin-2-yl)phenyl)carbamate(THKF-2).
Since Rf value of[18F]-THK523 and18F are 0.7 and 0.0, respectively,the above x/a was the count percentage of[18F]-THK523 to18F.In fact x/a is the labeling yield of[18F]-THK523,and it can be determined by TLC.Let P=x/a, Eq.(6)can be written as:
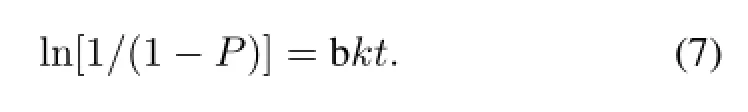
The plot of ln[1/(1−P)]vs.t shows a linear relationship and the rate constant k for the[18F]-THK523 formation can be calculated from the slope.
III.RESULTS AND DISCUSSION
There are several types of fluorinated precursor including the following substitute groups:(a)-OTs(tosylate),(b)-OTf (triflate),(c)-OMs(mesylate),and(d)-ONs(nosylate).In general,thereactivityisb>a>c>d,whilethestability for the groups is d>a>c>b.The tosylate was chosen for design of the[18F]-THK523 precursor because it compromises stability and reactivity.Figs.4 and 5 describe the synthesis of protected precursor THK-7 and cold standard of THKF-2. The synthesis of THK-7 involves the tosylation of a primary alcohol(THK-6).The Boc protection of the primary amino group in the intermediate THK-6 is important for a number of reasons.First,primary amino groups are generally morereactive towards tosyl chloride than primary alcohols.So,in the reaction conditions used to form precursor THK-7,it is expected that the amino group would be tosylated preferentially to the alcohol.Further,it is difficult to remove the aminotosyl protecting group by acid hydrolysis in a short reaction time and this would be detrimental for the radiolabelling reaction.
The choice of Boc as the protective group has the following advantages:Boc can be easily hydrolyzed by acid in a short reaction time and the by-products are CO2and iso-butanol(or isobutene),which can be easily evaporated.In addition,the presence of a free amino group in precursor BF241,used in Ref.[16],could lead to low radiosynthesis yield,as THK-6 may interact with K18F to form hydrogen bonds reducing the nucleophilicity of18F–.Fluorine is the most electronegative element.It can form hydrogen bonds with hydrogens on theamino group,while the Boc protected amino is a carbamate and it is less prone to the formation of hydrogen bonding. Comparing to the work of Fodero-Tavoletti et al.[16],the mostprominentadvantageofusingTHK-7,insteadofBF241, for radiolabeling of[18F]-THK523 was the improvement of radiochemical yields,as 40±5%and 24%(non-decay corrected)and at end of synthesis for THK-7 and BF241,respectively.There were no significant difference in radiochemical purity and specific activity.

TABLE 1.Mean labeling yields and specific radioactivity of[18F]-THK523 at different reaction time and temperature
Therefore,Boc protection of the primary amino group in the precursor,THK-7,is important to prevent side reactions, tosimplifyproductpurification,andtoimproveprecursorstability.
Radiolabelling yield of[18F]-THK523 was assayed by TLC.As shown in Table 1,the radiolabelling yield is relatively high,ranging from 11.91%to 75.11%(decay corrected to end of bombardment,EOB)and the synthesis time is 45–55min from EOB.
Radio-HPLC analysis showed the radiochemical purity (RCP)of[18F]-THK523 was≥90%.As shown in Fig.6, the retention time(tR)of[18F]-THK523 and precursor THK-7 were 6.12min and 12.94min,respectively.The chemical identity of[18F]-THK523 was verified by co-injection with the non-labelled standard THKF-2(tR=5.93min). The specific activity of[18F]-THK523 was determined as 2.5±0.5Ci/µmol(end of synthesis,EOS).
Labeling of radiopharmaceuticals is influenced by reaction time and temperature.Their relationship can be evaluated by chemical kinetics.Although TsO,as a leaving group during nucleophilic substitution,has been applied in many18F labeled radiopharmaceuticals,different molecular structures lead to different labeling yields.Evaluation of chemical kinetics is authentically necessary because of the importance of reaction temperature for labeling reaction.Labeling yields of[18F]-THK523 at different temperatures and minutes are listed in Table 1.The mean labeling yields and mean specific radioactivity increase with the temperature and time. At 120◦C,the rate constants were 4.95,4.97,5.00 and 5.04 for THK-7 contents of 5.24,26.2,52.4 and 131,respectively (Table 2).
For reaction chemical kinetics parameters,rate constant (k)increases with the increase of temperature,indicating that high temperature is conducive to increased reaction rate.In order to improve radiosynthesis yield,it is necessary to increase reaction temperature.However,[18F]-THK523 may be unstable or the binding of C-O or C-N may be broken down under such high temperature.

Fig.6.(Color online)HPLC identification of[18F]-THK523:analytical C18 column;80%ACN:20%H2O(0.05%triethylamine); flow 0.6mL/min.
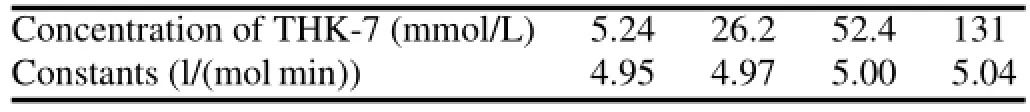
TABLE 2.Rate constants under different concentration of THK-7 (120◦C)
As shown in Table 1,labeling yields were high at 120◦C for reactions 15min and 30min(decay-corrected to EOB).Due to favorable thermal stability of[18F]-THK523 and for convenience in experiment operation,the optimal reaction temperature was chosen as 120◦C when labeling yields were larger than 70%at 15min. Calculations show the order of reaction is 1. The chemical kinetics study demonstrated the labeling reaction of 2-(2-(4-(tert-butoxycarbonyl)phenyl)quinolin-6-yloxy)ethyl 4-methylbenzenesulfonate(THK-7)was quite easily. This method can be applied to study labeling reactions of other radiopharmaceuticals.
The plot of ln[1/(1-P)]vs.t at four different concentration of THK-7 didn’t show significant linear relationship. The changesofrate constantsunderdifferentconcentration of precursor were shown in Table 2.The rate constant k didn’t change greatly with the concentration of THK-7.
The plot of ln[1/(1-P)]vs.t at different temperatures also showed a better linear relationship.The rate constant k increased with temperature,being 0.04,4.85 and 5.18 at 25◦C,120◦C and 160◦C,respectively.It demonstrated that k of the reaction was temperature dependent.From the variation of the order of magnitude,k had a significant increase from temperature 25◦C(0.04)to temperature 120◦C(4.95). These were the further evidence to validate the fact that temperature must be higher than 120◦C.
IV.CONCLUSION
[18F]-THK523 can be synthesized at CPCU with high synthesis yield(70±5%,n=6,decay-corrected to EOB).The total synthesis time was 45–55min and the average specific activity were 2.5±0.5Ci/µmol for[18F]-THK523(EOS)under optimum conditions.The chemical kinetics for[18F]-THK523 showed that the reaction order was 1 because concentration of precursor was much larger than that of18F.Low Eawas indicative of a low activation barrier.The fact that k increased with reaction temperature indicated that a rise of temperature favors an accelerated reaction rate.t1/2(the time for half of the reactant reacted)shortened with increased reaction temperature,which means that increasing temperature accelerates the labeling reaction.The protected precursor used in this method should be applicable for high yield automated production in a commercial synthesis module for clinical application in the future.The use of THK-7 might be suitable for routine production of high yield[18F]-THK523 and the radiosynthesis can be easily performed on an automated synthesis module.
[1]Wimo A and Winblad B.Handbook of Clinical Neurology.Elsevier,2008,89:137–146.
[2]McKhann G,Drachman D,Folstein M,et al.Neurology,1984, 34:939–939.
[3]Zhang W,Arteaga J,Cashion D K,et al.J Alzheimers Dis, 2012,31:601–612.
[4]Bulic B,Pickhardt M,Mandelkow E M,et al.Neuropharmacology,2010,59:276–289.
[5]Pritchard S M,Dolan P J,Vitkus A,et al.J Cell Mol Med, 2011,15:1621–1635.
[6]Aizenstein H J,Nebes R D,Saxton J A,et al.Arch Neurol-Chicago,2008,65:1509–1617.
[7]Lai M K,Chen C P,Hope T,et al.Neuroreport,2010,21: 1111–1115.
[8]Arriagada P V,Growdon J H,Hedley-Whyte E T,et al.Neurology,1992,42:631–639.
[9]VillemagneV,PikeK,DarbyD,etal.Neuropsychologia,2008, 46:1688–1797.
[10]Braskie M N,Klunder A D,Hayashi K M,et al.Neurobiol Aging,2010,31:1669–1778.
[11]Karran E,Mercken M,De Strooper B.Nat Rev Drug Discov, 2011,10:698–712.
[12]L˚angstr¨om B,Andr´en P E,Lindhe¨O,et al.Mol Imaging Biol, 2007,9:161–175.
[13]Matsumura K,Ono M,Yoshimura M,et al.Bioorg Med Chem, 2013,21:3356–3362.
[14]Matsumura K,Ono M,Hayashi S,et al.Med Chem Comm, 2011,2:596–600.
[15]Okamura N,Suemoto T,Furumoto S,et al.J Neurosci,2005, 25:10857–10962.
[16]Fodero-Tavoletti M T,Okamura N,Furumoto S,et al.Brain, 2011,134:1089–1100.
[17]Ono M,Hayashi S,Matsumura K,et al.ACS Chem Neurosci, 2011,2:269–275.
[18]Xia C F,Arteaga J,Chen G,et al.Alzheimers Dement,2013, 9:666–676.
[19]Matsumura K,Ono M,Kimura H,et al.ACS Med Chem Lett, 2011,3:58–62.
[20]Liu J,Kepe V,ˇZabjek A,et al.Mol Imaging Biol,2007,9: 6–16.
[21]Harada R,Okamura N,Furumoto S,et al.Eur J Nucl Med Mol I,2013,40:125–132.
[22]Cao G X,Zhou X Q,Liu Y T,et al.Nucl Sci Tech,2012,23: 52–56.
(
Received October 22,2013;accepted in revised form February 13,2014;published online August 6,2014)
10.13538/j.1001-8042/nst.25.040302
∗Supported by National Natural Science Foundation of China(Nos. 81271516 and 81371625),Program of Shanghai Science and Technology Commission(Nos.13JC1401503 and 14DZ1930402),the exchange program fund of doctoral student under the office for Graduate Medical Education,Fudan University and Shanghai Municipal Health and Family Planning Commission(No.2013313)
†Corresponding author,guanyihui@hotmail.com
猜你喜欢
Plasma Science and Technology(2023年9期)2023-10-08 08:20:54
Chinese Physics B(2022年11期)2022-11-21 09:39:38
Chinese Physics B(2022年4期)2022-04-12 03:47:38
作文小学中年级(2021年12期)2022-01-21 03:19:52
课堂内外·好老师(2021年2期)2021-09-12 08:23:17
医学前沿(2021年18期)2021-04-14 02:53:06
传奇·传记文学选刊(2020年6期)2020-06-29 07:42:27
作文周刊·小学一年级版(2016年4期)2016-08-11 12:58:13
知音(月末版)(2016年2期)2016-03-02 13:04:03
小小说月刊(2014年8期)2014-08-29 03:36:08
 Nuclear Science and Techniques2014年4期
Nuclear Science and Techniques2014年4期
- Nuclear Science and Techniques的其它文章
- Development of a beta radioluminescence nuclear battery∗
- Apparatus for determining permeability of hydrogen isotopes in molten-salt
- A facile method to build a proton nanosensor with neutral to basic pH sensitive range∗
- The flow characteristics of fluid in micro-channels of different shapes∗
- Hadron multiplicities in p+p and p+Pb collisions at the LHC∗
- An investigation on neutron induced reactions on stable CNO isotopes∗