
孤立条件下α-丙氨酸分子手性转变机制的密度泛函理论
2014-03-06 05:40王佐成刘凤阁赵衍辉于天荣
吉林大学学报(理学版) 2014年4期
王佐成,刘凤阁,吕 洋,赵衍辉,于天荣
(1.白城师范学院 物理学院,吉林 白城 137000;2.吉林大学 原子与分子物理研究所,长春 130012;3.白城医学高等专科学校 医学二系,吉林 白城 137000;4.白城师范学院 网络管理中心,吉林 白城 137000)
α-丙氨酸有左旋和右旋一对对映体,左旋体广泛存在于动物体内,对预防肾结石和协助葡萄糖代谢的作用较大,并有助于缓和低血糖[1];右旋体有抑菌作用,是自然保湿因子的主要成分,主要应用于手性药物、手性助剂和化妆品等领域[2].目前,关于手性α-丙氨酸分子的研究报道较多:如刘凤阁等[3]对α-丙氨酸分子手性对映体的结构特性进行了理论研究,龚䶮等[4]研究了丙氨酸对映体单晶变温偏振激光Raman光谱;王文清等[5]研究了α-丙氨酸分子的变温中子结构.本文通过对α-丙氨酸手性转变过程的研究,给出α-丙氨酸分子手性转变的反应路径及反应能垒,得到了α-丙氨酸分子手性转变的反应机制.
1 计算方法
本文在文献[3]的基础上将S型α-丙氨酸分子作为反应物,研究其到产物R型α-丙氨酸分子的过渡态[6-8]及中间体,并对包括过渡态在内的极值点前线分子轨道进行分析以获得分子的键特性.
计算基于密度泛函理论(DFT)的B3LYP[9-10]方法,采用双分裂价基选择基组,对C,O,N等原子加d极化函数,对H原子加p极化函数,即采用6-31+g(d,p)基组进行单重态势能面上的极小值、红外振动频率及前线分子轨道的理论计算.为验证过渡态的可靠性,对过渡态进行了内禀反应坐标(IRC)[11-14]分析.将过渡态和中间体等各极值点连接以确定反应路径.本文所有计算及图形均用Gaussian03/GaussView[15]软件完成.
2 结果与讨论
2.1 α-丙氨酸分子对映体的结构
在B3LYP/6-31+g(d,p)水平上的R型和S型α-丙氨酸分子的几何结构如图1所示.对于这种点手性分子对映体的转变,氢转移过程是最佳的反应途径[16].由图1可见,若实现对映体从S型到R型的手性转变,则需实现13H的位置移动.其过程会出现4C—1C—9C碳骨架的异构、4C与2H,3H,5H基团的异构、6N与7H,8H基团的异构以及13H—1C—6N键角对称改变的异构.
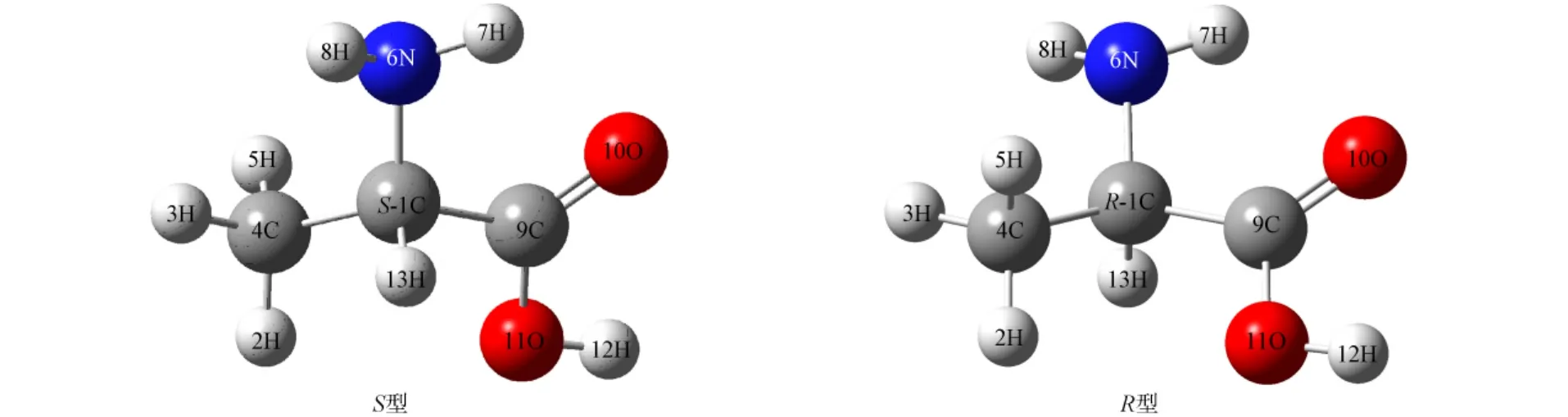
图1 在B3LYP/6-31+g(d,p)水平上的S型与R型α-丙氨酸分子的几何结构Fig.1 Geometries of(S)-and(R)-α-alanine molecules at the level of B3LYP/6-31+g(d,p)
2.2 α-丙氨酸分子手性转变路径的中间体及过渡态
对13H转移至10O上的结构在B3LYP/6-31+g(d,p)水平上进行几何结构优化、计算单重态的最低单点能和红外振动频率,并分析前线分子轨道,得到中间体(INT)的结构如图2(A)所示.计算结果显示,4C,1C,6N,9C,10O,11O,12H,13H基本处于同一平面上,二面角偏差均小于0.1°.能量为EINT(B3LYP)=-323.742 1a.u.,频率计算结果为无虚频,表明所得结构稳定.其前线分子轨道如图2(B)和(C)所示.INT的最高占据轨道(HOMO)主要来源于骨架原子p电子的贡献,骨架原子间1C与9C呈π键效应,其他原子的p电子呈局域特性.最低非占据轨道(LUMO)的骨架原子间及羧基的O和H间呈非键特性.在B3LYP/6-31+g(d,p)水平上分析过渡态TS1和TS2,计算过渡态的几何结构、能量、红外振动频率及其前线分子轨道.对过渡态进行全优化后的能量为:ETS1(B3LYP)=ETS2(B3LYP)=-323.649 5a.u.;红外振动均存在虚频:FreqTS1=-2 063.94cm-1,过渡态TS1和TS2的几何构型及虚频下的振动模式分别如图3和图4所示.

图2 在B3LYP/6-31+g(d,p)水平上计算的INT几何结构(A)与HOMO(B)和LUMO(C)前线分子轨道Fig.2 Calculated geometries of INT(A),frontier molecular orbitals HOMO(B)and LUMO (C)at the B3LYP/6-31+g(d,p)level
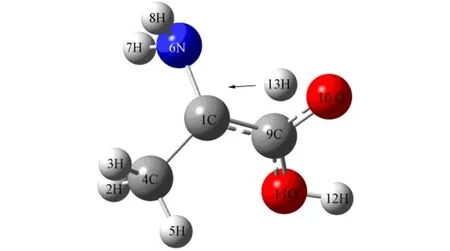
图3 TS1结构及虚频对应的振动模式Fig.3 Geometry and imaginary frequency vibrational mode of TS1
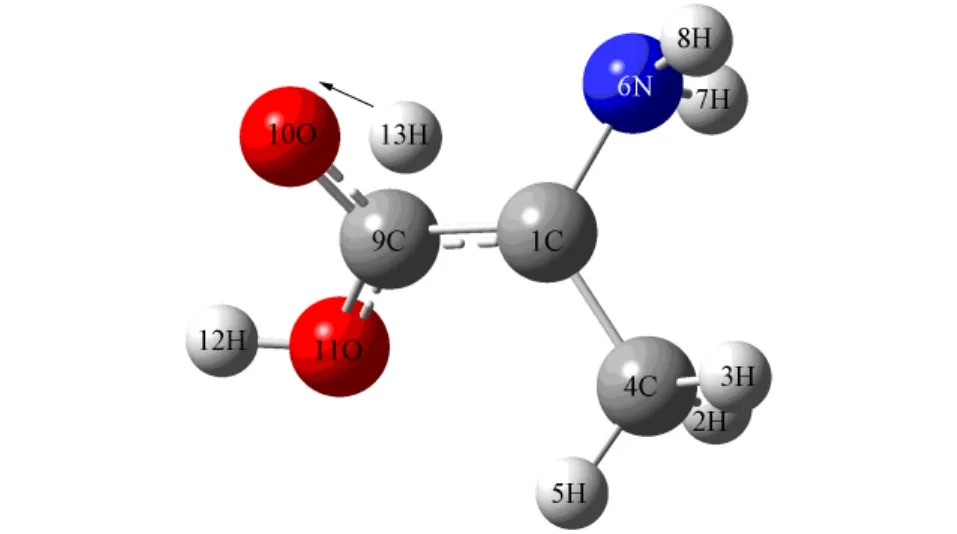
图4 TS2结构及虚频对应的振动模式Fig.4 Geometry and imaginary frequency vibrational mode of TS2
由图3可见,13H可从S型α-丙氨酸分子的1C迁移至10O上,该过程伴随碳氮骨架、6N基团与羧基的异构,形成INT.由图4可见,13H可从INT的10O上迁移至R型α-丙氨酸分子的1C上,该过程也伴随碳氮骨架、6N基团与羧基的异构,形成R型α-丙氨酸分子.TS1和TS2的HOMO及LUMO分别如图5和图6所示.
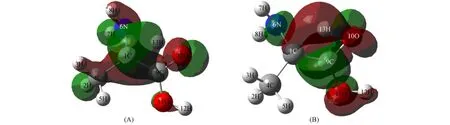
图5 TS1的HOMO(A)及LUMO(B)Fig.5 HOMO(A)and LUMO(B)of TS1
由图5和图6可见:HOMO轨道均为10O原子的p电子与13H原子的s电子贡献,具有明显的反键特征;LUMO轨道为1C和10O原子的p电子与13H的s电子贡献,具有成键键特征.因此,TS1和TS2是分别连接反应物与中间体及中间体与生成物的过渡态.
2.3 α-丙氨酸分子手性转变过渡态的IRC计算
为验证过渡态的可靠性,即过渡态TS1为连接S型对映体与INT,TS2为连接INT与R型对映体,分别对TS1和TS2在B3LYP/6-31+g(d,p)理论水平上进行IRC路径探测,结果如图7所示.
由图7(A)可见,其能量最高点为过渡态TS1,最低点分别为中间体INT和S型α-丙氨酸分子的结构,优化后得到中间体和S型α-丙氨酸分子结构.由图7(B)可见,其能量最高点为过渡态TS2,最低点分别为R型α-丙氨酸分子和中间体INT,优化后分别得到中间体和R型α-丙氨酸分子的结构.S型对映体转变为中间体的过程如图8所示.中间体转变为R型对映体的过程如图9所示.
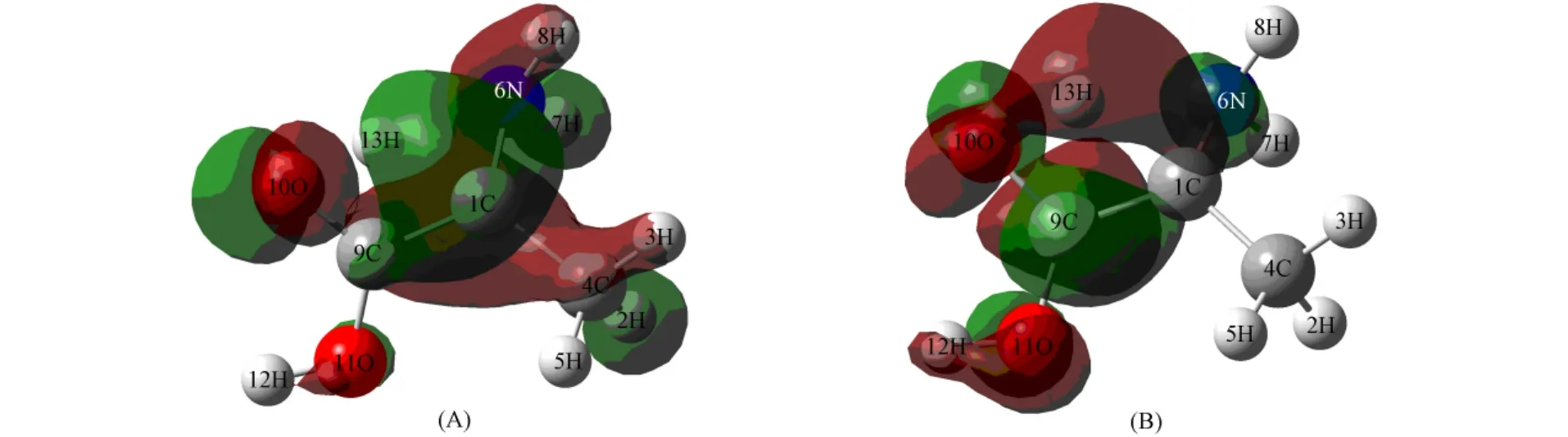
图6 TS2的HOMO(A)和LUMO(B)Fig.6 HOMO(A)and LUMO(B)of TS2
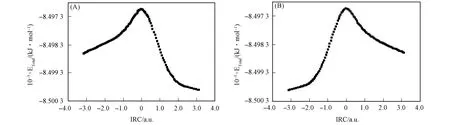
图7 B3LYP/6-31+g(d,p)理论水平上对过渡态TS1(A)和TS2(B)进行的IRC分析Fig.7 IRC analysis for transition state TS1(A)and TS2(B)at the level of B3LYP/6-31+g(d,p)
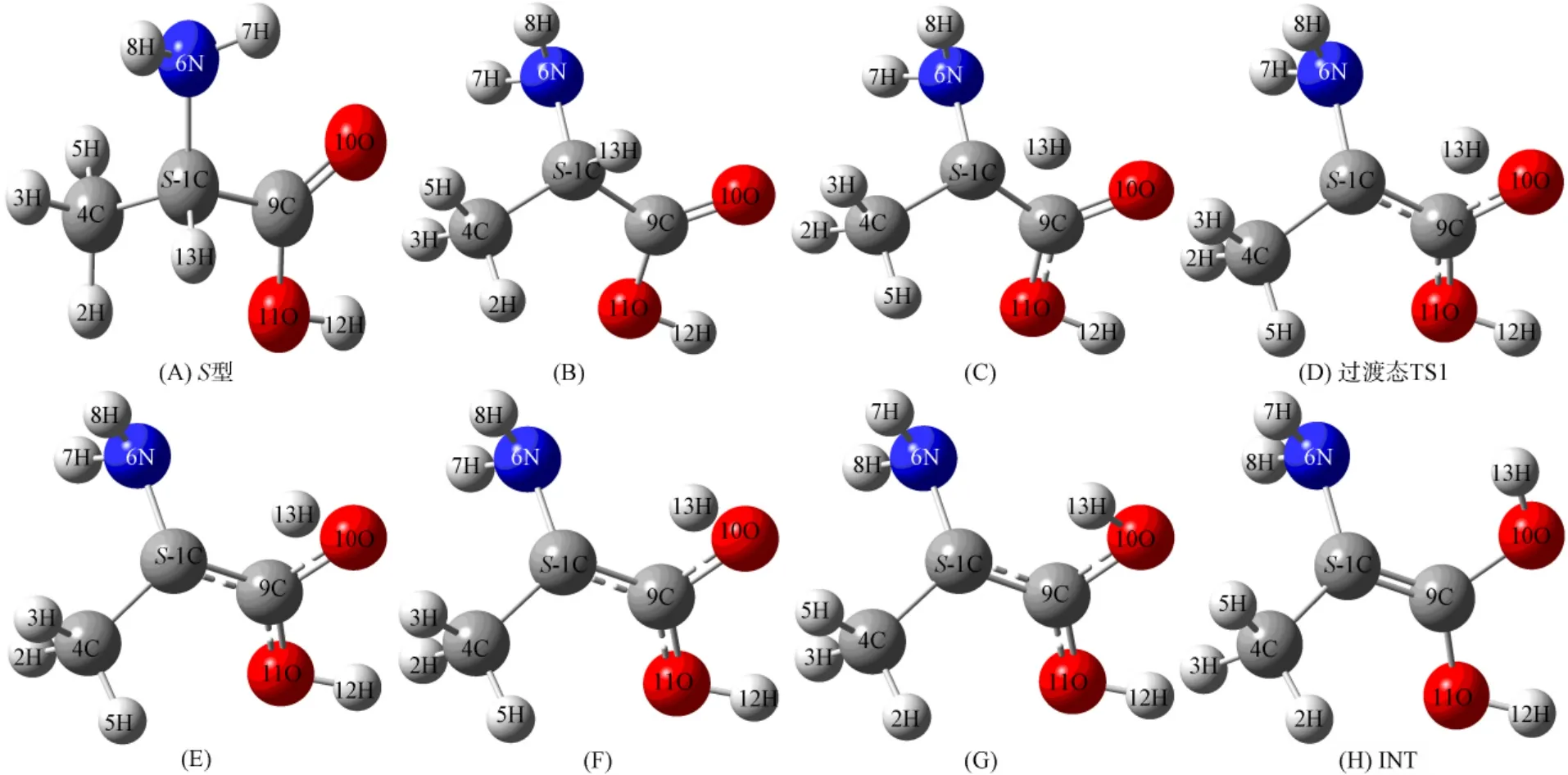
图8 S型对映体转变为中间体的过程Fig.8 Conversion process from (S)-enantiomer into intermediates
2.4 α-丙氨酸分子手性转变路径
综上可知,α-丙氨酸分子从S型到R型的手性转变路径为S→TS1→INT→TS2→R,反应路径中各状态结构的主要几何参数列于表1,稳定点与过渡态的能量列于表2.由表2可见,S型α-丙氨酸分子经过渡态TS1转变至中间体INT需跨越的能垒为326.6kJ/mol.实现此过程需要外界干预.中间体INT再跨越1个243.1kJ/mol的能垒,即可由过渡态TS2转变至R型α-丙氨酸分子,实现手性对映体转变.完成手性转变所对应的势能面示意图如图10所示.

图9 中间体转变为R型对映体的过程Fig.9 Conversion process from intermediates into(R)-enantiomer

表1 反应路径中各稳定点及过渡态的主要几何参数*Table 1 Principal geometric parameters of stable points and transition states in pathway

表2 在B3LYP/6-31+g(d,p)水平上计算的反应路径中稳定点及过渡态的能量和相对能量Table 2 Energies,relative energies of each stable point and transition state of pathways calculated at the B3LYP/6-31+g(d,p)level
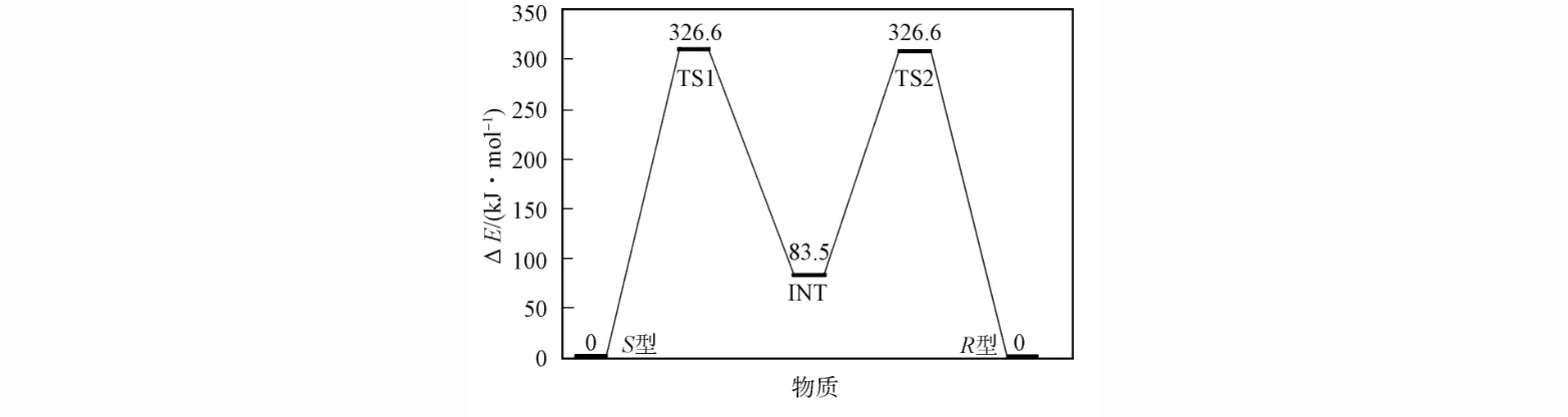
图10 在B3LYP/6-31+g(d,p)水平上计算的α-丙氨酸分子手性转变过程势能面示意图Fig.10 Potential surfaces diagram ofα-alanine molecule chiral transition process calculated at the B3LYP/6-31+g(d,p)level
综上,本文使用DFT的B3LYP方法,采用6-31+g(d,p)基组,计算了α-丙氨酸分子从S型到R型的手性转变过程.结果表明,该反应路径为:先由S型α-丙氨酸分子跨越1个326.6kJ/mol的能垒达到过渡态TS1,TS1经过1个放热过程转变为中间体INT后,再跨越1个243.1kJ/mol的能垒,经过渡态TS2转变至R型α-丙氨酸分子,经过1个放热过程,即可实现手性对映体转变.由于2个能垒均较高,因此该手性转变过程需在一定的外界条件干预下才能实现.
[1]赵亚华.分子生物学教程 [M].北京:科学出版社,2011:8-15.(ZHAO Yahua.Molecular Biology Tutorial[M].Beijing:Science Press,2011:8-15.)
[2]林国强,李月明,陈耀全,等.手性合成不对称反应及其应用 [M].北京:科学出版社,2003:15-21.(LIN Guoqiang,LI Yueming,CHEN Yaoquan,et al.Asymmetric Reactions of Chiral Synthesis and Its Application[M].Beijing:Science Press,2003:15-21.)
[3]刘凤阁,赵衍辉,钱研,等.孤立条件下手性α-丙氨酸分子结构特性的理论研究 [J].吉林师范大学学报:自然科学版,2013(4):47-51.(LIU Fengge,ZHAO Yanhui,QIAN Yan,et al.Theoretical Study on Structural Characteristics of Chiral Alpha Alanine Under Isolated Condition[J].Journal of Jilin Normal University:Natural Science Edition,2013(4):47-51.)
[4]龚䶮,易芳,王文清.丙氨酸对映体单晶的变温偏振激光拉曼光谱研究 [J].光散射学报,2002,14(3):145-149.(GONG Yan,YI Fang,WANG Wenqing.Temperatur Dependent Polarized Raman Study ofD-andL-Alanine Single Crystals[J].Chinese Journal of Light Scattering,2002,14(3):145-149.)
[5]WANG Wenqing,LIU Yinan,GONG Yan,et al.Parity Violations on Molecular Chirality:Neutron Crystal-Structure ofD-and-L-Alanine[J].Acta Phys-Chim Sin,2004,20(11):1345-1351.
[6]Eyring H.The Activated Complex and the Absolute Rate of Chemical Reaction[J].Chemical Reviews,1935,17(1):65-77.
[7]Garrett B C,Truhlar D G.Generalized Transition State Theory.Classical Mechanical Theory and Applications to Collinear Reactions of Hydrogen Molecules[J].Journal of Physical Chemistry,1979,83(8):1052-1079.
[8]Garrett B C,Truhlar D G.Criterion of Minimum State Density in the Transition State Theory of Bimolecular Reactions[J].The Journal of Chemical Physics,1979,70(4):1593-1598.
[9]Becke A D.Density-Functional Thermochemistry.Ⅲ.The Role of Exact Exchange[J].J Chem Phys,1993,98:5648-5652.
[10]徐光宪.量子化学 [M].北京:科学出版社,1999:95-112.(XU Guangxian.Quantum Chemistry[M].Beijing:Science Press,1999:95-112.)
[11]Fukui K.Formulation of the Reaction Coordinate [J].The Journal of Physical Chemistry,1970,74(23):4161-4163.
[12]Gonzalez C,Schlegel H B.An Improved Algorithm for Reaction Path Following[J].The Journal of Chemical Physics,1989,90:2154-2161.
[13]Gonzalez C,Schlegel H B.Reaction Path Following in Mass-Weighted Internal Coordinates [J].Journal of Physical Chemistry,1990,94(14):5523-5527.
[14]Ishida K,Morokuma K,Komornicki A.The Intrinsic Reaction Coordinate.AnabinitioCalculation for HNC→HCN and H-+CH4→CH4+H-[J].The Journal of Chemical Physics,1977,66(5):2153-2156.
[15]Frisch M J T,Schlegel H B,Scuseria G E,et al.Gaussian 03,Revision D.01 [CP/OL].Pittsburgh,PA:Gaussian Inc,2003.
[16]TIAN Chuanjin,XIU Peng,MENG Yan,et al.Enantiomerization Mechanism of Thalidomied and the Role of Water and Hydroxide Ions[J].Chem:A Eur J,2012,18(45):14305-14313.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
大学化学(2021年8期)2021-09-26
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
电脑知识与技术(2018年3期)2018-03-21
中成药(2017年9期)2017-12-19
哈尔滨理工大学学报(2017年1期)2017-04-08
医药导报(2015年6期)2015-02-10
郑州大学学报(理学版)(2013年2期)2013-03-11
