
镍基催化剂对衣康酸连续化催化加氢研究
2014-03-04 10:04陶军宏李小年江大好
化工生产与技术 2014年2期
陶军宏 李小年 江大好
(浙江工业大学工业催化研究所,杭州 310032)
甲基丁二酸具有手性,可用于废纸漂白、蚀刻剂、酯化反应、光化学反应,是合成大量的手性化合物和药物大分子的不可或缺的试剂[1-2]。传统工业合成采用酯水解法,以丁烯酸为原料生成巴豆酸乙酯,通过多步水解、提取和重结晶制备甲基丁二酸,过程复杂、操作步骤多、成本高、收率较低[3]。
衣康酸具有α,β-不饱和羰基的结构,具有活泼的化学性质,可进行自身间的聚合,是一种生物质原料[4]。用于不饱和羧酸选择性加氢较多的是钌系催化剂以及铑系等贵金属催化剂[5-9]。Sinou D等用水溶性铑络合物[Rh(cod)Cl](cod=1,5-环辛二烯)对PhCH=C(COOH)NHAc进行α,β-碳碳双键的还原结果表明,在323 K下,HCOONH4水溶液中反应15 h,水溶性铑络合物[Rh(cod)Cl]可以有效地选择性催化碳碳双键获得65%的转化率和43%的选择性[10]。近年来,负载型镍基催化剂以其价廉、活性高等优点在多种物质的加氢中得到了广泛的应用[11-13]。
本研究以衣康酸为原料,在固定床上用Ni/SiO2催化剂一步连续化催化加氢合成甲基丁二酸,具有一步合成、成本低、收率高、三废少、有利于环境保护等优点,是一条很有希望放大进行工业化的工艺路线。考察了Ni负载量、反应温度和反应压力对衣康酸连续化催化加氢的影响,优化了反应条件,并通过BET、XRD和TEM对催化剂进行了表征。
1 实验部分
1.1 催化剂制备
Ni/C催化剂由浸渍法制备。将一定量的蒸馏水加入培养皿中,按相应Ni负载量 (质量分数5%~20%)加入Ni(NO3)2·6H2O溶液,混匀之后倒入称好的10 g二氧化硅(筛孔0.38~0.83 mm),搅拌均匀后用吹风机迅速吹干,待无液体流动时,将表面皿置于烘箱内,于空气中100℃下烘24 h后,转移至坩埚于马弗炉程序升温至450℃焙烧4 h,即得Ni/SiO2催化剂。催化剂采用原位还原,于氢气中400℃还原2 h,H2体积流量20mL/min。
1.2 催化剂表征
催化剂的BET测试在Nova 1000e型孔结构比表面积测试仪上进行。样品加热至523 K,恒温12 h后进行脱气预处理,以除去样品上的吸附物种。在77 K下由N2物理吸附法进行测定,比表面积采用BET方程由氮气吸附等温线求得,分别以Barrett-Joyner-Halenda(BJH)和Horvath-Kawazoe(HK)模型测得样品的中孔和微孔结构。
在Thermo SCINTAG X"TRA型X-射线衍射仪(XRD)上进行XRD分析,采用Cu Ka辐射,管压为45 kV,管电流为40mA,波长0.154 056 nm,固体探测器,以谢乐(Scherrer)公式推导出晶粒大小。
采用Tecnai G2 F30 S-Twin型300 kV高分辨透射电子显微镜(TEM)获得样品形貌,工作电压为300 kV。将样品粉末研磨于乙醇中超声分散后支撑于铜网上,干燥后测定。
1.3 催化剂评价
催化剂评价采用在气固反应固定床中进行。在气固反应固定床反应管中装入筛孔0.38~0.83 mm的催化剂0.5 g,通入氢气进行催化剂原位还原后,用恒流泵将原料溶液,即衣康酸在乙醇中的质量浓度为0.125 g/mL,以0.05 mL/min的体积流量经一定温度预热后进入固定床反应管,氢气作为载气和反应原料,体积流量为20 mL/min,恒压3 MPa反应一段时间后,取样,减压蒸馏,减压蒸馏温度为50~120℃,烘干。
反应方程式如下:
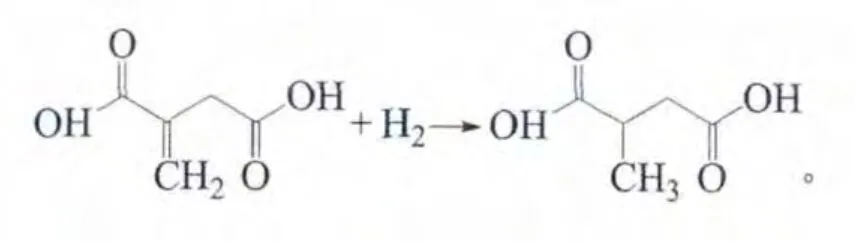
2 结果与讨论
2.1 催化剂的N2物理吸附分析
图1是SiO2和质量分数分别为5%的Ni/SiO2、10%的Ni/SiO2、15%的Ni/SiO2的N2吸附-脱附等温线。

图1 SiO2、5%Ni/SiO2、10%Ni/SiO2、15%Ni/SiO2的N2吸附-脱附等温线Fig 1 N2 adsorption-desorption isotherm of SiO2, 5%Ni/SiO2,10%Ni/SiO2,15%Ni/SiO2
从图1可以看出,不同镍负载量的SiO2催化剂样品均具有介孔的典型特征,即出现IV型吸脱附曲线,并且均带有H1型迟滞环。负载金属后并没有改变载体SiO2的介孔结构。在相对压力<0.1,吸附形式一部分以单层吸附的方式发生在微孔的表面,还有一部分以单层和多层吸附的形式发生在介孔的内部孔壁上。相对压力为0.6~1.0时,SiO2呈现出的迟滞环为H1型,其程度与位置表明了介孔孔径的分布范围。由图1还可知,金属负载前后迟滞环发生了一定的变化,这说明金属的负载对SiO2的介孔结构还是产生了一定程度的影响。
表1是不同SiO2和Ni/SiO2的结构参数。
由表1可以看出,未负载的SiO2载体的比表面积为289.3m2/g,总孔容为1.168 cm3/g;负载Ni金属后,催化剂的比表面积和孔容均有所下降,这可能是负载的NiO存在于载体SiO2的表面或者进入孔道内部引起的。
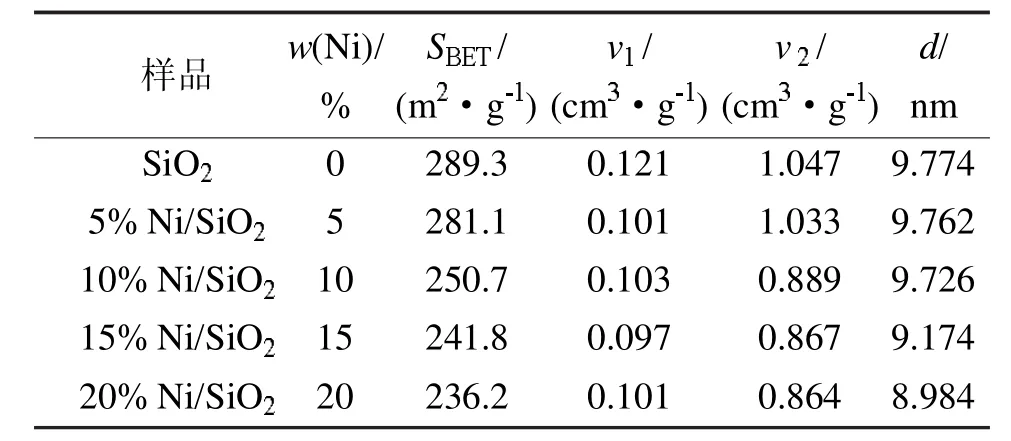
表1 不同SiO2和Ni/SiO2的结构参数Tab 1 Structure parameters of different SiO2 and Ni/SiO2
2.2 催化剂的XRD分析
图2是不同负载量镍催化剂的XRD。

图2 不同负载量的Ni/SiO2的XRD图谱Fig 2 XRD patterns of catalysts
从图2可以明显看到4个峰。第1个为非晶态SiO2的弥散特征峰(2θ=22°),后3个为NiO晶相特征衍射峰(2θ分别为37.3°、43.3°、62.9°),没有发现活性组分镍与载体SiO2作用生成的新物相,这表明镍与载体之间的相互作用较弱,NiO主要以游离态的形式存在于载体表面上。随着负载量的上升,峰的尖锐程度增加,由线宽法和系勒公式,说明随着负载量的增加,镍的晶粒度变大。
2.3 催化剂的TEM分析
图3给出了不同负载量的Ni/SiO2催化剂的TEM显微照片。
在TEM下直接观察,可以发现随着负载量的增加,Ni的分散度变差,存在比较明显的聚积的现象,这可能是透射过程中产生的,和实际催化剂镍物种的分散略有差异,这个表征结果和前面的XRD结果相一致。

图3 不同负载量的Ni/SiO2的TEM照片Fig 3 TEM micrographs of nikel-based catalysts
2.4 催化剂的活性评价结果
不同Ni含量的Ni/SiO2催化剂对衣康酸连续化选择性催化加氢的影响如表2所示。

表2 不同Ni含量Ni/SiO2催化剂对催化加氢的影响Tab 2 Effectof Ni contenton selective hydrogenation of itaconic acid
由表2可以看出,Ni负载质量分数分别为5%、10%、15%、20%的催化剂所对应衣康酸的转化率依次为60.3%、70.4%、75.2%、80.6%,即衣康酸的加氢转化率随镍含量增多显著升高。选择性还原C=C键所得产物甲基丁二酸的选择性由 73.5%增至88.1%。通常,金属含量增加,金属粒子的粒径也会随着增大。
2.5 反应温度对衣康酸催化加氢的影响
表3给出了温度对衣康酸选择性加氢生成甲基丁二酸的影响。
从表3可看出,随着反应温度升高,衣康酸的转化率显著增加,且甲基丁二酸选择性在30~90℃时变化不大,略有下降。当温度继续升高至110℃,C=C键选择性大幅降低,副产物含量却明显升高。原因可能是温度升高到一定温度后,衣康酸本身发生聚合生成一些低聚副产物,导致甲基丁二酸的选择性降低。

表3 反应温度对催化加氢反应的影响Tab 3 Effectof temperature on selective hydrogenation of itaconic acid
由此可知,提高反应温度存在2方面的影响:1)温度升高会提高衣康酸选择性加氢反应的反应速率,但温度升高又可能引发副反应的发生,生成不必要的副产物,使目标产物甲基丁二酸的选择性受影响而降低;2)温度升高,H2在溶液中的溶解度降低,对加氢反应造成不利影响,部分抵消了温度对反应速率的促进效应。
2.6 反应压力对衣康酸催化加氢的影响
表4给出了压力对衣康酸选择性加氢生成甲基丁二酸的影响。

表4 反应压力对催化加氢反应的影响Tab 4 Effectof pressure on selective hydrogenation of itaconic acid
从表4可看出,对于一个液相催化加氢反应,随着反应压力的增加,原料衣康酸的转化率从65.8%显著提高到85.4%,这是因为增加氢气的压力,可以使液相中氢气的含量增加,从而增加催化剂表面氢气的吸附量,加快反应速度。产物甲基丁二酸的选择性由81.3%提高至88.1%,但是随着压力的继续增加,甲基丁二酸的选择性迅速降低至50.2%。在反应压力为5 MPa的时候,衣康酸具有α,β-不饱和羰基的结构,具有活泼的化学性质,原料衣康酸在高压下大量进行自身间的低聚合,造成了高转化率低选择性的结果。
3 结论
研究制备的一系列不同Ni负载量的Ni/SiO2都有很高的比表面积和很大孔径,Ni负载量的增加,镍的晶粒度变大,应提高镍的分散性,在对衣康酸的连续化催化加氢反应中表现了很好的加氢活性和选择性,在氢气压力为3 MPa、反应温度为70℃时,衣康酸的转化率可达80.6%,甲基丁二酸的转化率为88.1%。
[1]Brzyska W,Debska E,Szczotka M.New complexes of rare earth elementswithmethylsuccinic acid[J].Polish Journal of Chemistry,2011,75(10):1393-1400.
[2]Gelman F,Avnir D,Schuman H,et al.Sol gel entrapped chiral rhodium and ruthenium complexes as recyclable catalysts for the hydrogenation of itaconic acid[J].Journal of Molecular Catalysis A,Chemical,1999,146(1):123-128.
[3]Takafumi A,Fumio T,Hiroyuki N,et al.Pumice-supported Cu-Pd catalysts:influence of copper on the activity and selectivityofpalladium in thehydrogenationofphenylacetylene and butlebe[J].Journal of Catalysis,1999,182(2):456-462.
[4]Achiwa K.Highly enantioselective catalytic asymmetric hydrogenation of itoaconic acid[J].Tetrahedron Letters,1978, 19(17):1475-1476.
[5]Tao Y H,Kang R H,Ou Yang X M.Recent development inthe selective reduction of α,β-unsaturated carbonyl compound[J].Chin Org Chem,2000,20(4):441-453.
[6]Maki-Arvela P,Hajek J,Salmi T,et al.Chemoselectivehydrogenation of carbonyl compoundsoverheterogeneouscatalysts [J].Applied Catalysis A:General,2005,292(18):1-49.
[7]Zhang Yuankui,Liao Shijian,Xu Yun.Catalytic selective hydrogenation of cinnamaldehyde to hydrocinnamaldehyde[J].Applied Catalysis A:General,2000,192(14):247-251.
[8]NHoller H,Lin W M.Activity and selectivity of Ni-Cu/ Al2O3catalysts for hydrogenation of crotonaldehyde and mechanism of hydrogenation[J].Journal of Catalysis,1984,85(1):25-30.
[9]Gu M M,Zhang S Y,Wang L F.Studies on selective catalytic hydrogenation;I.Selective heterogeneous catalytic hydrogenation ofα,β-unsaturated aldehyde and ketones[J].Chin Org Chem,1984,4(6):454-456.
[10]Sinou D,San M,Claver C,et al.Chiral sulphonated phosphines.Part VII.Catalytic transfer-hydrogenation of unsaturated substrateswith formates in the presence ofwater soluble complexes of rhodium[J].JMol Catal,1991,68:L9-L12.
[11]Pawelec B,Daza L,Fierro J L G,et al.Regeneration of Ni-USY catalysts used in benzene hydrogenation[J].Appl Catal A,1996,145(1):307-308.
[12]Rautanen P A,Aittamaa JR,Krause A O I.Liquid phase hydrogenation of tetralin on Ni/Al2O3.Chem.Eng.Sci.,2001, 56(4):1247-1251.
[13]Suh D J,Park T J,Lee SH,Kim K L.Nicke-l alumina composite aaerogels as liquid-phase hydrogenation catalysts [J].JNon-Cryst Solids,2001,285(1/3):309-310.
猜你喜欢
化学工程师(2023年1期)2023-02-17
上海塑料(2022年4期)2022-08-29
机械工业标准化与质量(2022年6期)2022-08-12
理化检验-化学分册(2020年12期)2021-01-26
上海农业科技(2019年1期)2019-02-22
中国果业信息(2018年5期)2018-01-17
中国调味品(2017年2期)2017-03-20
中国塑料(2016年1期)2016-05-17
中国塑料(2016年11期)2016-04-16
中国塑料(2016年7期)2016-04-16
