
线粒体功能障碍和氧化应激在脂肪肝中的作用机制
2014-03-03 05:46:06李志凌卿笃信
分子诊断与治疗杂志 2014年4期
李志凌 卿笃信
·综述·
线粒体功能障碍和氧化应激在脂肪肝中的作用机制
李志凌 卿笃信★
目前各种病因引起的肝脏脂肪变性甚至发展为脂肪性肝炎或脂肪性肝硬化成为当今严重的公共卫生及医疗问题。氧化应激、NO信号通路的中断和线粒体功能障碍等被认为是加速脂肪变性和启动脂肪肝和纤维化进程的关键机制。线粒体损伤和氧化应激之间相互作用的复杂机制,也使得临床治疗脂肪性肝炎效果不佳。因此找到一种多基因、多靶点安全有效阻断氧化应激和线粒体损伤的分子靶向药物和治疗方案成为了学术界的难题。
线粒体功能障碍;线粒体损伤;氧化应激;脂肪肝
各种病因引起的肝脏脂肪变性甚至发展为脂肪肝成为当今严重的公共卫生及医疗问题。脂肪酸和甘油三酯异常蓄积是肝细胞发生脂肪变性的首要步骤。当机体受到“二次打击”时,肝细胞进一步破坏。另外,氧化应激、线粒体功能障碍、一氧化氮(nitric oxide,NO)信号通路的中断等被认为是加速脂肪变性和导致脂肪肝的关键机制。
Ludwig等[1]发现肥胖、2型糖尿病及每天少于20 g乙醇饮酒量人群的肝脏出现脂肪变性,在病理组织结构上的变化与酒精性脂肪肝炎相同,故首次提出非酒精性脂肪性肝炎(nonalcoholic steatohepatitis,NASH)的概念。NASH与胰岛素抵抗相关的肥胖、2型糖尿病、高脂血症等因素有关,而在酒精性脂肪肝炎(alcoholic steatohepatitis,ASH)中也出现胰岛素抵抗(insulin resistance,IR)的现象,但是其中的病理机制并未完全明确[2_4]。1998年Day等[5]提出的“二次打击”假说阐明了长期酒精饮用、IR、肝细胞脂肪变性与线粒体功能障碍之间的关系,并阐明了线粒体损伤和功能障碍是作为NASH和ASH发展的共同途径。
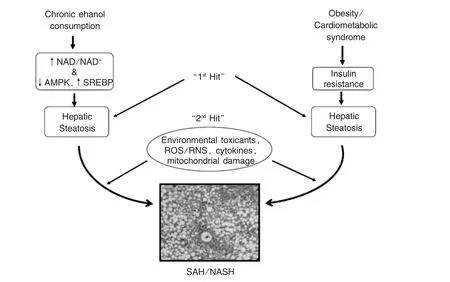
图1 ASH和NASH形成的共同途径[6]Figure 1 Pathophysiology of alcoholic steatohepatitis(ASH)and nonalcoholic steatohepatitis(NASH)[6]
1 ASH和NASH的“一次打击”假说
1.1ASH的“一次打击”假说
乙醇代谢的过程中,线粒体中NADH/NAD+比例增高,脂肪酸的酯化作用增强,合成更多的甘油三酯。同时肝脏对脂肪酸的β氧化能力下降,极低密度脂蛋白(very low density lipoprotein,VLDL)的合成能力下降,最终导致脂质在肝脏中的积聚引起肝细胞的脂肪变性。这个过程即为ASH的一次打击。相关流行病学调查发现酒精对胰岛素及其信号通路的影响存在剂量效应关系,即低剂量起到保护作用,而长期大剂量可损害肝脏中的胰岛素信号通路[7]。有些研究者发现长期大量饮酒能够引起IR,可导致肝细胞中脂肪的积累,但是缺乏支持的证据[8]。
1.2NASH的“一次打击”假说
研究发现IR在NASH患者群中占较高的比例[9]。多数学者认为胰岛素信号转导的改变和脂肪代谢的失衡是脂肪肝形成的主要启动因素[10]。胰岛素具有抑制激素敏感性脂肪酶,发生IR时外周脂肪组织分解加强[11]。同时,IR可导致脂蛋白脂肪酶基因的过度表达,使得游离脂肪酸(free fatty acid,FFA)增多。FFA由脂肪酸转运蛋白转运或扩散及摄取作用进入肝脏[12]。在高脂饲养的脂肪酸转运蛋白5基因敲除的动物模型中肝脏脂肪变性减少、空腹血糖和胰岛素水平降低[13]。FFA还刺激胰岛素的信号转导抑制因子3表达增加而加重IR。已发生脂肪沉积的肝细胞表面胰岛素受体减少且出现胰岛素受体酪氨酸磷酸化,进一步加重IR和促进非酒精性脂肪性肝病发生[14]。此外,升高的胰岛素将增加载脂蛋白B100的分解和肝型脂肪酸结合蛋白基因缺失均可引起VLDL分泌降低,脂类物质易蓄积于肝脏,引起肝细胞脂肪变性[15]。在多个研究中发现瘦素抵抗与胰岛素抵抗有关系[16_17]。因此,推测胰岛素抵抗和瘦素抵抗是NASH第一次打击的重要原因。这提示胰岛素抵抗和脂肪肝的发生的关系还需进一步研究。
2 “二次打击”假说
肝脏发生脂肪变性后饮食、吸烟、毒物或药物及代谢因素(高血糖、高甘油三酯、高胆固醇血症等)等二次打击因素,促使肝脂肪变性向脂肪性肝炎进展。二次打击通过线粒体缺陷,增加肝脏中氧化应激的产物如活性氧(reactive oxygen species,ROS)和活性氮,促使肝脏由单纯脂肪变性进一步发展为脂肪性肝炎[18]。目前认为氧化应激和线粒体功能障碍在脂肪肝“二次打击”中起了关键作用。下文将详细说明氧化应激和线粒体与脂肪肝之间的关系。
3 线粒体功能缺陷在脂肪肝中的作用
线粒体是肝细胞最重要的细胞器之一,是脂肪酸进行β-氧化和三羧酸循环、腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)合成和ROS形成的主要场所[19]。有些学者发现脂肪酸产生直接脂毒性,可造成线粒体肿胀,降低线粒体内酶的活性,引起肝细胞线粒体功能障碍,进而诱导肝细胞凋亡[20]。脂肪酸在肝细胞线粒体氧化过程中,一小部分电子可漏溢而直接与氧反应形成超氧阴离子自由基和其他ROS[21]。而在胆碱诱导线粒体缺陷的小鼠中观察到脂肪肝形成也证实了线粒体在脂肪肝形成中的重要性[22]。
3.1氧化应激
细胞呼吸利用氧合成ATP,由氧衍生的ROS的产生及其作用超过对其防御或清除的能力时,可造成组织、细胞损伤的状态称为氧化应激。ROS包括羟基(OH_)、超氧阴离子基(O2_)、过氧化氢(H2O2)等。氧化应激主要来自于线粒体脂肪酸的氧化和过氧化反应、细胞色素P450 2E1[23]和细胞色素P450 4A对长链和超长链脂肪酸的ω-氧化,以及各种原因引起线粒体复合体Ⅰ和Ⅲ的分子缺陷[24]。硬脂酰辅酶A去饱和酶1的减少[25]及抗氧化物质耗尽也可导致ROS的蓄积。在使用抗氧化药物硫辛酸的小鼠实验中,发现其能够阻止高脂肪饮食对小鼠肝脏的影响,调节参与脂肪生成和线粒体内β-氧化的基因及提高胰岛素的敏感性,并能诱导线粒体增加和解偶联蛋白-2(uncoupling protein 2,UCP-2)基因的表达[26]。
ROS还能激活氧化还原反应敏感性激酶,进而激活抑制性Kappa B激酶β(inhibitor kappa B kinaseβ,IKKβ),IKKβ可诱导转录因子Kappa B(nuclear transcription factor Kappa B,NF-κB)的产生。NF-κB可提高肿瘤坏死因子α和白介素-8的水平,两者可进一步促进ROS的形成,也可以诱导多形核粒细胞的趋化,进而又加强分泌TNFα和白介素-8,导致局部炎症细胞大量浸润,引起肝脏的炎症反应[27]。在ob/ob小鼠动物实验中使用抗-肿瘤坏死因子抗体处理四周后,能够降低肝脏中的脂肪含量及下调IKKβ和NF-κB水平,减缓非酒精性脂肪性肝病的进展[28]。故作在酒精性和非酒精性脂肪肝中脂质过氧化和作为ROS主要来源的线粒体中起了重要作用。
3.2线粒体与氧化应激
在血清内FFA增多或乙醇代谢过程中刺激ROS大量生成。当超过抗氧化物质的清除能力而导致ROS蓄积时,ROS把线粒体作为首要的打击靶子,直接损害线粒体膜DNA,线粒体膜DNA编码的呼吸链复合体亚基水平降低[29]和核糖体缺陷引起的线粒体蛋白合成系统发生障碍[30],导致肝细胞脂肪代谢失调和电子在呼吸链流动中阻断而被传递到分子氧产生ROS,最终导致线粒体功能障碍甚至肿胀、破裂。同样在胆碱缺乏饮食诱导的小鼠和ob/ob小鼠的肝脏均浆中均检测到呼吸复合体活性降低[31]。ROS可以通过氧化线粒体膜上的心磷脂而影响呼吸链复合体酶的活性[32],引起ROS生成增多及ATP合成减少。早期脂肪变性的肝脏往往表现为线粒体超微结构损伤[33]。但在脂肪肝和肥胖的小鼠模型中,通过检测分离线粒体和肝脏均浆中的氧消耗、ATP合成情况等,发现实验组中的肥胖小鼠和对照组中的瘦体质小鼠线粒体功能并无差别[34]。提示线粒体损伤可能和脂肪变性可能存在剂量效应关系,目前相关研究尚不明确。
3.3线粒体与脂质过氧化
过多FFA沉积在肝细胞,引起细胞色素酶P450 2E1表达明显增高,ROS增多引起氧化应激,最终导致细胞坏死[35]。生成过多的ROS与线粒体生物膜的磷酯、酶和膜受体相关多聚不饱和脂肪酸的侧链及核酸等大分子物质反应形成脂质过氧化产物如丙二醛和壬烯,其能够与线粒体蛋白结合和进一步损害线粒体呼吸链复合物[36],直接或间接的损害线粒体DNA,诱导线粒体产生更多ROS形成恶性循环[37]。多聚不饱和脂肪酸过氧化产物还可促进载脂蛋白B100溶解,而减少VLDL的合成,加重甘油三酯在肝脏内的积聚。同时多聚不饱和脂肪酸过氧化产物具有中性粒细胞趋化作用,能诱导肝小叶内中性粒细胞浸润,产生炎症反应甚至引起肝细胞坏死[38]。同时还可激活kupffer细胞和星状细胞促进肝纤维化甚至发生肝硬化[39]。脂质过氧化反应还可消耗细胞内和线粒体内的抗氧化物,导致ROS清除减少,进一步加重氧化应激。在脂肪肝小鼠模型中通过补充n-3多不饱和脂肪酸如二十碳五烯酸和二十二碳六烯酸,能够刺激脂肪代谢和抑制脂肪生成,而起到保护作用[40]。因此氧化应激与脂质过氧化相互作用共同影响线粒体功能,促进脂肪肝性肝炎的形成。
3.4线粒体与UCP-2
持续的氧化应激状态诱导了线粒体功能的调整,NF-κB的激活同时可诱导部分抗氧化蛋白的表达,如刺激线粒体UCP-2表达增加,UCP-2是一种位于线粒体内膜上的载体蛋白,可以调节线粒体膜电位和ROS的产生,可增加内膜的质子漏过率,进一步影响ATP合成[41]。线粒体中UCP-2表达上升为一种适应性反应,UCP-2使ATP合成减少抑制脂质合成,而且ATP合成解偶联,可降低线粒体4期呼吸的氧化还原压力,限制ROS的产生,减少脂质氧化,保护肝细胞及线粒体免受损伤[42]。在高脂肪饮食饲养的动物模型中通过白藜芦醇诱导UCP-2的上调,发现其线粒体含量较高脂肪饮食的对照组有明显增高,并且未观察到肝脏炎症反应,达到保护肝脏作用[43]。但UCP-2使线粒体ATP合成受抑制,在肝脏短暂缺血、能量需求急剧增加、应激等情况下ATP供给不足时,肝细胞对外界打击更加敏感[44]。细胞内ATP的减少也抑制了细胞周期素依赖性激酶和细胞周期素依赖性激酶抑制因子的功能,这些综合作用将导致受损肝细胞的再生功能下降,影响了细胞的修复,也另一面加速了应激状态下肝脏病变的进展。
3.5线粒体与一氧化氮
活性氮,主要包括过氧亚硝基阴离子(ONOO_)及其质子形式过氧亚硝酸(HOONO)等具有高度氧化活性的自由基和硝基类化合物。体内的一氧化氮(NO)由NO合成酶催化产生。在健康的肝脏中,可诱导型一氧化氮合酶水平非常低或是无法检测到的。长期饮酒或肥胖时,炎症细胞浸润和通过诱导kupffer细胞、肝细胞和胆管上皮细胞使得可诱导型一氧化氮合酶的表达增加,这可能与线粒体蛋白的硝化作用有关[45]。NO扩散进入线粒体,抑制细胞色素c氧化酶,与O2_形成过氧化亚硝酸盐(ONOO_)。NO和过氧亚硝基阴离子(ONOO_)能够介导线粒体功能缺陷[46],NO通过与细胞色素C氧化酶亚铁血红素位点可逆性结合来调节线粒体氧化呼吸活性和通过与可溶性鸟苷酸环化酶相互作用[47],导致ATP合成减少,ROS生成增加,肝细胞对缺氧应激的适应能力降低,加重肝脏损伤[48]。
4 小结
随着“一次打击”和“二次打击”假说的提出,氧化应激和线粒体功能障碍在脂肪肝病发病机制中的作用日益受到学者们的重视,大量的研究逐渐开展,线粒体损伤和氧化应激之间相互作用的机制被广大学者认可,但在线粒体损伤中的各种影响因素之间存在着潜在的联系且相互作用,相互促进,互为因果,造成理论应用到临床治疗上出现巨大的难题。目前,国内外在治疗脂肪肝并无特效的药物和治疗方案,大多从单一的作用途径着手,如给予抗氧化的药物、改善线粒体功能的药物、降低细胞色素P450 2E1含量的药物,这些药物均能在一定程度上降低氧化应激损伤,延缓线粒体损伤,改善病理生理指标,但无法逆转氧化应激和线粒体损伤,因此找到一种多基因、多靶点安全有效阻断氧化应激和线粒体损伤的分子靶向药物和治疗方案,是我们近阶段将要面临的挑战。
[1]Ludwig J,Viggiano T R,McGill D B,et al.Nonalcoholic steatohepatitis:Mayo Clinic experiences with a hitherto unnamed disease[J].Mayo Clin Proc, 1980,55(7):434_438.
[2]Albert van de Wiel A.Diabetes mellitus and alcohol[J].Diabetes Metab Res Rev,2004,20(4):263_267.
[3]BunoutD.Nutritionalandmetaboliceffectsof alcoholism:their relationship with alcoholic liver disease [J].Nutrition,1999,15(7_8):583_589.
[4]Howard A A,Arnsten J H,Gourevitch M N.Effect of alcohol consumption on diabetes mellitus:a systematic review[J].Ann Intern Med,2004,140(3):211_219.
[5]Day C P.Non-alcoholic fatty liver disease:a massive problem[J].Clin Med,2011,11(2):176_178.
[6]Mantena S K,King A L,Andringa K K,et al.Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol-and obesity-induced fatty liver diseases[J].Free Radic Biol Med,2008,44(7):1259_ 1272.
[7]He L,Simmen F A,Mehendale H M,et al.Chronic ethanol intake impairs insulin signaling in rats by disrupting Akt association with the cell membrane.Role of TRB3 in inhibition of Akt/protein kinase B activation[J].J Biol Chem,2006,281(16):11126_11134.
[8]He L,Marecki J C,Serrero G,et al.Dose-dependent effectsofalcoholoninsulinsignaling:partial explanation for biphasic alcohol impact onhuman health[J].Mol Endocrinol,2007,21(10):2541_2550.
[9]Asrih M,Jornayvaz F R.Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance[J].J Endocrinol,2013,218(3):R25_R36.
[10]Gaggini M,Morelli M,Buzzigoli E,et al.Nonalcoholicfattyliverdisease(NAFLD)andits connectionwithinsulinresistance,dyslipidemia, atherosclerosis and coronary heart disease[J].Nutrients, 2013,5(5):1544_1560.
[11]Lomonaco R,Ortiz-Lopez C,Orsak B,et al.Effect of adiposetissueinsulinresistanceonmetabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease[J].Hepatology,2012, 55(5):1389_1397.
[12]Zhou J,Febbraio M,Wada T,et al.Hepatic fatty acid transporter Cd36 is a common target of LXR,PXR, andPPARgammainpromotingsteatosis[J].Gastroenterology,2008,134(2):556_567.
[13]Falcon A,Doege H,Fluitt A,et al.FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase[J].Am J Physiol Endocrinol Metab,2010,299(3):E384_E393.
[14]Machado M V,Ferreira D M,Castro R E,et al.Liver and muscle in morbid obesity:the interplay of fatty liver and insulin resistance[J].PLoS One,2012,7(2): e31738.
[15]Ibrahim S H,Kohli R,Gores G J.Mechanisms of lipotoxicity in NAFLD and clinical implications[J].J Pediatr Gastroenterol Nutr,2011,53(2):131_140.
[16]Wang T N,Chang W T,Chiu Y W,et al.Relationships between changes in leptin and insulin resistance levels in obese individuals following weight loss[J].Kaohsiung J Med Sci,2013,29(8):436_443.
[17]Zain S M,Mohamed Z,Mahadeva S,et al.Impact of leptin receptor gene variants on risk of non-alcoholic fatty liver disease and its interaction with adiponutrin gene[J].J Gastroenterol Hepatol,2013,28(5):873_879.
[18]Begriche K,Igoudjil A,Pessayre D,et al.Mitochondrial dysfunction in NASH:causes,consequences and possible means to prevent it[J].Mitochondrion,2006,6 (1):1_28.
[19]Morris E M,Rector R S,Thyfault J P,et al.Mitochondria and redox signaling in steatohepatitis[J].Antioxid Redox Signal,2011,15(2):485_504.
[20]Kusminski C M,Shetty S,Orci L,et al.Diabetes and apoptosis:lipotoxicity[J].Apoptosis,2009,14(12): 1484_1495.
[21]McKenzie M,Liolitsa D,Hanna M G.Mitochondrial disease:mutations and mechanisms[J].Neurochem Res, 2004,29(3):589_600.
[22]Aharoni-Simon M,Hann-Obercyger M,Pen S,et al.Fatty liver is associated with impairedactivityof PPARgamma-coactivator 1alpha(PGC1alpha)and mitochondrial biogenesis in mice[J].Lab Invest,2011, 91(7):1018_1028.
[23]Bansal S,Liu C P,Sepuri N B,et al.MitochondriatargetedcytochromeP4502E1inducesoxidative damage and augments alcohol-mediated oxidative stress [J].J Biol Chem,2010,285(32):24609_24619.
[24]Fromenty B,Robin M A,Igoudjil A,et al.The ins and outsofmitochondrialdysfunctioninNASH[J].Diabetes Metab,2004,30(2):121_138.
[25]Feldstein A E,Bailey S M.Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease[J].Antioxid Redox Signal,2011,15(2):421_ 424.
[26]Valdecantos M P,Perez-Matute P,Gonzalez-Muniesa P,et al.Lipoic acid administration prevents nonalcoholic steatosis linked to long-term high-fat feeding by modulating mitochondrial function[J].J Nutr Biochem, 2012,23(12):1676_1684.
[27]Kleniewska P,Piechota-Polanczyk A,Michalski L,et al.Influence of block of NF-kappa B signaling pathway on oxidative stress in the liver homogenates [J].Oxid Med Cell Longev,2013,2013:308358.
[28]Li Z,Yang S,Lin H,et al.Probiotics and antibodies to TNFinhibitinflammatoryactivityandimprove nonalcoholic fatty liver disease[J].Hepatology,2003, 37(2):343_350.
[29]Albano E.Oxidative mechanisms in the pathogenesis of alcoholic liver disease[J].Mol Aspects Med,2008,29 (1_2):9_16.
[30]Weiser B,Gonye G,Sykora P,et al.Chronic ethanol feeding causes depression of mitochondrial elongation factorTuintheratliver:implicationsforthe mitochondrial ribosome[J].Am J Physiol Gastrointest Liver Physiol,2011,300(5):G815_G822.
[31]Petrosillo G,Portincasa P,Grattagliano I,et al.Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I,reactive oxygen species and cardiolipin[J].Biochim Biophys Acta, 2007,1767(10):1260_1267.
[32]Musatov A.Contribution of peroxidized cardiolipin to inactivation of bovine heart cytochrome c oxidase[J].Free Radic Biol Med,2006,41(2):238_246.
[33]Kojima H,Sakurai S,Uemura M,et al.Mitochondrial abnormalityandoxidativestressinnonalcoholic steatohepatitis[J].Alcohol Clin Exp Res,2007,31(1 Suppl):S61_S66.
[34]Flamment M,Arvier M,Gallois Y,et al.Fatty liver and insulin resistance in obese Zucker rats:no role for mitochondrial dysfunction[J].Biochimie,2008,90(9): 1407_1413.
[35]Garcia-Ruiz C,Baulies A,Mari M,et al.Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance:Cause or consequence[J].Free Radic Res,2013,47(11):854_868.
[36]Landar A,Zmijewski J W,Dickinson D A,et al.Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species[J].Am J Physiol Heart Circ Physiol,2006,290(5):H1777_H1787.
[37]Begriche K,Igoudjil A,Pessayre D,et al.MitochondrialdysfunctioninNASH:causes, consequences and possible means to prevent it[J].Mitochondrion,2006,6(1):1_28.
[38]Tilg H,Diehl A M.Cytokines in alcoholic and nonalcoholic steatohepatitis[J].N Engl J Med,2000, 343(20):1467_1476.
[39]Lieber C S.Alcoholic fatty liver:its pathogenesis and mechanism of progression to inflammation and fibrosis [J].Alcohol,2004,34(1):9_19.
[40]Espinosa A,Valenzuela R,Gonzalez-Manan D,et al.Preventionofliversteatosisthroughfishoil supplementation:correlation of oxidative stress with insulin resistance and liver fatty acid content[J].Arch Latinoam Nutr,2013,63(1):29_36.
[41]Guan L L,Wang Y F,Gong D Z,et al.[Establishment of the Chang liver cell line stably overexpressing human UCP2 gene and its effect on mitochondrial membrane potential and reactive oxygen species][J].Zhonghua Gan Zang Bing Za Zhi,2012,20(2):131_ 135.
[42]Serviddio G,Bellanti F,Tamborra R,et al.Uncoupling protein-2(UCP2)induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis(NASH)liver to ischaemia-reperfusion injury[J].Gut,2008,57(7):957_965.
[43]Poulsen M M,Larsen J O,Hamilton-Dutoit S,et al.Resveratrol up-regulates hepatic uncoupling protein 2 and prevents development of nonalcoholic fatty liver disease in rats fed a high-fat diet[J].Nutr Res,2012, 32(9):701_708.
[44]Serviddio G,Bellanti F,Tamborra R,et al.Uncoupling protein-2(UCP2)induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis(NASH)liver to ischaemia-reperfusion injury[J].Gut,2008,57(7):957_965.
[45]Garcia-Ruiz I,Rodriguez-Juan C,Diaz-Sanjuan T,et al.Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice[J].Hepatology,2006, 44(3):581_591.
[46]Eccleston H B,Andringa K K,Betancourt A M,et al.Chronic exposure to a high-fat diet induces hepatic steatosis,impairs nitric oxide bioavailability,and modifies the mitochondrial proteome in mice[J].Antioxid Redox Signal,2011,15(2):447_459.
[47]Nisoli E,Tonello C,Cardile A,et al.Calorie restriction promotes mitochondrial biogenesis by inducing the expressionof eNOS[J].Science,2005,310(5746):314_ 317.
[48]Shiva S,Oh J Y,Landar A L,et al.Nitroxia:the pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway[J].Free Radic Biol Med,2005,38(3):297_306.
The mechanisms of mitochondrial dysfunction and oxidative stress in fatty liver disease
LI Zhiling,QING Duxin★
(Gastroenterology Department,The Second Xiangya Hospital of Central South University,Hunan,Changsha 410011,China)
Currently hepatic steatosis,steatohepatitis and fatty liver cirrhosis have become a serious public health and medical problem.Oxidative stress,nitric oxide(NO)signaling disruption,and mitochondrial dysfunction are proposed to be key mechanisms that accelerate steatosis and initiate progression to steatohepatitis and fibrosis.The complex mechanisms of the interaction between mitochondrial damage and oxidative stress lead to poor treatment effects for steatohepatitis.As a result,it has become an academic problem to find a safe and effective multi-gene treatment that can block the procedure of oxidative stress and mitochondrial damage.
Mitochondrial dysfunction;Mitochondrial damage;Oxidative stress;Fatty liver disease
湖南省科技厅课题(06FJ4086)
中南大学湘雅二医院消化内科,湖南,长沙410011
★通讯作者:卿笃信,E-mail:qingdx@163.com
猜你喜欢
中老年保健(2022年5期)2022-11-25 14:16:14
中国造纸(2022年9期)2022-11-25 02:24:54
中老年保健(2022年1期)2022-08-17 06:14:08
昆明医科大学学报(2022年2期)2022-03-29 00:52:18
保健医苑(2021年7期)2021-08-13 08:47:54
家庭医学(下半月)(2020年7期)2020-08-24 07:47:04
环球时报(2019-04-03)2019-04-03 04:15:14
幸福·健康版(2016年10期)2016-11-17 11:21:46
癌变·畸变·突变(2016年3期)2016-02-27 06:15:36
哈尔滨医药(2015年4期)2015-12-01 03:57:54
