
施万细胞与背根节神经元体外共培养成髓鞘模型的建立*
2014-02-22 03:38:14苏文凤韦中亚沈筠恬
交通医学 2014年4期
苏文凤,韦中亚,顾 芸,沈筠恬,陈 罡
(南通大学江苏省神经再生重点实验室,江苏226001)
施万细胞与背根节神经元体外共培养成髓鞘模型的建立*
苏文凤**,韦中亚,顾 芸,沈筠恬,陈 罡
(南通大学江苏省神经再生重点实验室,江苏226001)
目的:探讨施万细胞与背根节神经元髓鞘化共培养的标准化方法,为研究周围神经髓鞘化的形成机制提供稳定的周围神经髓鞘化体外模型。方法:取出生1~3 d新生SD大鼠,培养施万细胞,经纯化鉴定后用于共培养。取孕14~15 d的SD大鼠胚鼠背根神经节,经纯化后用于共培养;将2种分别纯化的细胞进行共培养,加抗坏血酸诱导髓鞘的形成。利用免疫组化染色、扫描电镜和透射电镜,检测髓鞘的形成。结果:纯化后施万细胞纯度可达到98%以上,可用于共培养。背根节神经元贴壁良好,经纯化后几乎无杂细胞可见,可用于共培养。对共培养细胞进行MAG与NF的免疫组化染色,发现有数量可观的髓鞘形成,并且髓鞘是紧密包绕在神经元轴突上。扫描电镜下可观察到施万细胞包绕轴突形成髓鞘,而透射电镜下则可观察到有致密的髓鞘板层结构形成。结论:本实验成功建立稳定可靠的体外成髓鞘模型,可用于轴突的新髓鞘化实验研究。
施万细胞;背根节神经元;髓鞘形成;免疫荧光染色法;大鼠
1 材料与方法
1.1 材料 (1)实验动物:SPF级出生1~3d的SD大鼠培养施万细胞,SPF级孕14~15d的SD大鼠培养背根节神经元。大鼠(均由南通大学实验动物中心提供,动物使用许可证:XYSK(苏)2007-0021),实验过程中对动物处置符合动物伦理学标准。(2)主要试剂:神经生长因子(NGF),5-氟尿嘧啶(5-FU),尿嘧啶 (U),0.25%胰酶,Forskolin,L-抗坏血酸,ITS supplement,Thy 1.1和β-阿糖胞苷均(Sigma公司);HRG (Human neuregulin)β1(proteintech公司);DMEM培养基,Neurobasal培养基(NB),B27,胎牛血清(FBS,Gibco公司);补体(Invitrogen公司);兔抗MAG,鼠抗NF-200,鼠抗S100(Sigma公司);羊抗兔IgG-Cy3,羊抗鼠IgG-Cy3和羊抗鼠IgG-Alex-488(Invitrogen公司)。
1.2 方法 (1)施万细胞的培养与纯化:出生1~3d的新生SD大鼠,经冰冻、75%乙醇消毒后,无菌条件下取出坐骨神经。剥膜后放入1mg/mL胶原酶和0.125%的胰酶消化30min,以1×106个/mL的密度接种于预先用PDL包被的培养皿中。放入37℃,5%的CO2培养箱中,24h后换成含β-阿糖胞苷(终浓度为10μmol/L)的DMEM培养基,培养24h后换成终浓度为2μmol/L Forskolin和10 ng/mL LHRGβ1的10% FBS DMEM培养基,每3d换液。3~5d待细胞生长至融合,消化收集细胞悬液,加成纤维细胞特异性抗体Thy 1.1(1∶1 000稀释于10%FBS DMEM培养基)。冰上孵育2h,离心弃上清,加补体(1∶3稀释于DMEM培养基)重悬,37℃孵育1h。DMEM培养基清冼2遍后,接种于预先PDL包被的培养皿中备用。(2)背根节神经元的培养与纯化:孕14~15d的SD大鼠,通风橱内乙醚麻醉后备皮。75%的乙醇消毒后,无菌条件下取出胚鼠,解剖镜下取出背根神经节,将背根神经节接种于PDL包被的24孔板中。24h后换含有U(10μmol/L)、5-FU(10μmol/L)、Glutamine(2mmol/L)和2%B27的Neurobasal培养基培养2d。后换含有Glutamine(2 mmol/L)和2%B27的Neurobasal培养基培养2d,重复前面步骤3次,即可得到较纯的背根节神经元。以上所使用的培养基均含有50ng/mL的NGF。(3)背根节神经元与施万细胞共培养:将纯化后的施万细胞培养1~2d,0.125%的胰酶消化,制备单细胞悬液后计数。细胞密度调至1×105个/mL,以500μL/孔的量与事先纯化好的神经元细胞共培养。1d后换成含有Glutamine(2 mmol/L)和2%B27的Neurobasal培养基培养2d。2d后换成含有2%ITS和2%BSA的DMEM培养基培养4d(每2d换1次液)。然后换成含 50μg/mL抗坏血酸 15%FBS DMEM培养基进行培养,直到髓鞘形成(每2d半量换液)。以上所使用的培养基均含有50ng/mL的NGF。(4)免疫荧光染色:施万细胞纯度鉴定,经过纯化后的施万细胞,用0.01 mol/L PBS洗3遍后,4%多聚甲醛室温下固定30min。0.01 mol/L PBS清冼3遍,加入适量封闭液室温封闭1h,然后加入鼠抗S100(1∶400)一抗4℃孵育过夜。0.01 mol/L PBS清冼3遍,每次5min,避光加入羊抗鼠IgG-Cy3(1∶1 000)二抗室温下避光孵育2h。弃掉二抗进行Hochest室温染色15min,于激光共聚焦显微镜下拍照观察。MAG免疫荧光染色:施万细胞与背根节神经元共培养21d后,用0.01mol/L PBS洗3次,4%多聚甲醛室温固定10min,弃掉4%多聚甲醛用预冷的甲醇-20℃固定6min。弃掉甲醇后用0.01M PBS洗3次,封闭液室温封闭1h。然后用兔抗MAG(1∶200)一抗和鼠抗NF(1∶500)一抗4℃孵育过夜,0.01mol/L PBS洗3次。避光加入羊抗兔IgG-Cy3(1∶1 000)二抗和羊抗鼠IgG-488(1∶1 000)二抗,室温孵育1h。用0.01 mol/L PBS洗3次后,于激光共聚焦显微镜下拍照观察。(5)扫描电镜:细胞经处理后,固定于预冷的2.5%的戊二醛。鞣酸洗涤后固定于1%锇酸,乙醇梯度脱水,叔丁醇置换2次,放入冷冻干燥机冷冻干燥后粘贴标本并喷金,扫描电镜下观察标本。(6)透射电镜:细胞经处理后,固定于预冷的2.5%的戊二醛,后再固定于1%锇酸,梯度乙醇脱水,Epon812环氧树脂包埋,半薄切片定位,超薄切片(70nm),枸橼酸铅和醋
酸铀染色。透射电镜下观察标本。
2 结 果
2.1 施万细胞的培养与纯化 利用施万细胞和成纤维细胞2种细胞的增殖周期、贴壁速度及表面抗原不同,首先利用药物除去部分增殖较快的成纤维细胞,进行初步纯化。待细胞增殖生长至融合后,仍有大量成纤维细胞存在(图1A),再利用抗体-补体分别结合除去成纤维细胞,进行深度纯化。经过以上2步纯化,几乎无杂细胞可见,且细胞排列整齐方向一致(图1B),S100免疫组化染色结果显示施万细胞的纯度可达到98%以上(图1C-E),可用于后续实验研究。
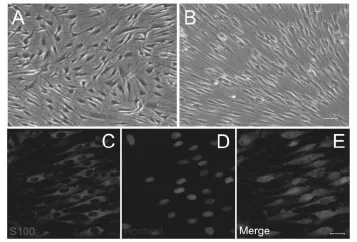
图1 施万细胞纯化前后相差显微镜下对比及纯化后纯度鉴定
2.2 背根节神经元的培养与纯化 从孕14~15 d的SD大鼠胚鼠中取背根神经节,用抗有丝分裂试剂尿嘧啶和5-氟尿嘧啶除去植块中增殖的细胞,如成纤维细胞、施万细胞等。如图2显示了背根神经节从贴壁1 d~16 d后背根节神经元的纯化状态,在纯化了16 d后基本上无杂细胞存在,可用于后续研究。

图2 相差显微镜下观察背根节神经元的纯化过程
2.3 细胞共培养 将纯化后的施万细胞制成悬液,以105个/mL的密度接种在含背根节神经元的玻片上(图3A)。贴壁过夜后换成无血清培养基,此时施万细胞开始增殖以及调整与轴突的方向(图3B),为成髓鞘做准备。1周后施万细胞的增殖达到一定的数目,然后用含有抗坏血酸的培养基进行诱导培养。约1周后,在相差显微镜下可观察到颜色较深的“条索状”物(图3C)。随着培养时间的延长,在相差显微镜下可观察到更多的“条索状”物形成(图3D)。扫描电镜下可观察到施万细胞包绕神经元的轴突形成髓鞘(图5A、C)。培养21 d后,对共培养细胞进行MAG与NF的免疫组化染色(图4A-F),结果显示神经元轴突上紧密包绕了数量可观的髓鞘,经透射电镜检测发现形成致密的板层结构(图5B、D)。
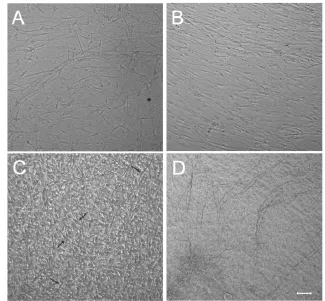
图3 相差显微镜下观察施万细胞与背根节神经元共培养的过程

图4 共培养21d时髓鞘蛋白MAG免疫荧光染色

图5 电镜下观察共培养成髓鞘的结构
3 讨 论
髓鞘化过程在体内受到神经、免疫、脉管、内分泌系统等多种系统的调节。多种影响因素共同作用的结果,导致体内周围神经髓鞘化的微环境极其复杂,严重影响了研究的进度和可靠性。因此,建立稳定可靠地体外髓鞘模型是研究成髓鞘机制的重要技术手段。当今大鼠背根节神经元和施万细胞的共培养模型已被用于各种实验研究中[9-11],几乎所有的共培养都是分为2种细胞分别纯化和纯化后2种细胞的共培养过程。培养过程中会遇到诸多问题,主要有包被基质的选择和包被时间、纯化试剂的选择和纯化步骤、共培养培养基、添加试剂及因子的选择和作用时间等。
本实验研究结合多种不同的培养方法加以改进,充分利用已有的研究基础,建立了一个规范稳定的体外成髓鞘培养模型。首先是包被基质的选取,合适的包被结构可以为背根节神经元突起的生长提供一个稳定的外环境。包括Matrigel(基质胶)以及laminin(层粘连蛋白)的应用。Matrigel虽有报道可作为包被基质,为神经元的生长提供一个良好的环境[6]。但它也存在许多不足之处:⑴换液时基质胶很容易被吸起导致脱落;⑵基质胶与塑料皿底的结合能力很弱;⑶不同批次的基质胶质量差别较大,因此重复性较差;⑷基质胶在37℃培养15d后,与培养皿的结合不稳定,从而很容易脱离下来。Laminin对背根节神经元突起的生长和神经元的存活较有利[7],因为神经元体外培养的贴壁能力较差,很容易飘浮而死亡,严重干扰了实验的进行。为了增加神经元的稳定性,很多学者在使用Laminin包被之前要先用
PLL(左旋)包被[8]。这种包被方式较繁琐费时,且有文献报道Laminin影响髓鞘的形成[12],因此不利于一些实验的研究。本实验采用对细胞毒性较小的PDL (右旋)进行包被,对于24孔板中背根节神经元的稳定性较好,且操作简单影响因素单一。
其次是施万细胞的纯化。施万细胞纯化的一个主要困难是大量的成纤维细胞的掺杂,对于怎样得到纯度较高的施万细胞,现在已有很多方法进行了报道。例如,利用2种细胞的增殖周期不同、贴壁速度不同以及表面抗原不同等[4,13-14]。本实验结合各方法的利弊在组织消化时间、因子作用时间及纯化步骤上加以改进,可得到纯度达到98%以上的施万细胞(图1B-E所示)。
再者是背根节神经元的纯化。据大鼠胚胎的发育特点,我们取孕14~15 d的SD大鼠胚胎鼠的背根神经节。不管是在体内还是体外,在这个时期大约有5%的成神经细胞仍具有分裂能力,且大部分背根节神经元仍没有到达对应的靶组织[8]。此时背根节含有神经元、成纤维细胞、施万细胞、吞噬细胞和各种各样的其他类型的细胞,这些细胞除神经元外均具有较强的分裂增殖能力。因此可用阿糖胞苷、尿嘧啶和5-氟尿嘧啶等抗有丝分裂试剂进行除去[15]。我们分别比较2组有丝分裂抑制剂对增殖细胞以及神经元的影响。结果发现,阿糖胞苷组增殖细胞很快被除去,作用约1周后杂细胞基本不可见,但神经元的活力大大降低,与尿嘧啶和5-氟尿嘧啶组相比差很多,以致慢慢死去。尿嘧啶和5-氟尿嘧啶组约2周后杂细胞基本不可见(图2),且神经元突起较长、活力较好,可用于后续实验研究。但神经元的贴壁能力相比于施万细胞要差很多,轻微的晃动或换液时液体冲击力过大都可能导致神经元的漂浮,因此,在移动培养板或换液时要尽可能轻柔。
最后是将2种纯化后的细胞共培养。当施万细胞与背根节神经元的轴突接触时,两者可相互作用首先引起施万细胞的大量增殖[16]。体外共培养的条件下,施万细胞和背根节神经元必须达到合适的密度才能引起施万细胞的增殖[6]。因此本实验24孔板我们采用105个/mL密度进行接种,刚贴壁的细胞零散分布,经过短暂的调整开始大量增殖且方向一致、排列整齐(图3)。在此培养过程中,我们分别更换不同的培养基使施万细胞与神经元处于最佳生长环境。其中B27和ITS的加入,它们包含有很多因子,可以促进胶质细胞的分化和髓鞘的形成。例如胰岛素、孕酮、转铁蛋白和硒盐的加入[5],孕酮可激活髓鞘蛋白P0和PMP的启动[17],且可增加P0蛋白和MBP的表达水平[18]。神经营养因子对神经元的发育至关重要,它可以控制细胞的存活、分化、突起生长、兴奋性、突触可塑性和功能发挥等[19]。NGF对轴突信号的控制起重要作用,它可以通过TrkA信号通路控制髓鞘的形成[20],因此整个神经元培养过程中都有NGF的加入。抗坏血酸可诱导髓鞘的形成,在未加入抗坏血酸时,可见髓鞘蛋MAG有表达但无其他髓鞘蛋白的表达,且不能形成髓鞘[21-22]。本实验中,当施万细胞增殖到一定数目,加入抗坏血酸约1周,可看到“条索状”物形成(图3C),免疫组化结果显示此“条索状”物为形成的髓鞘,随着时间的延长“条索状”物大量出现。同时我们也在扫描电镜下观察到施万细胞包绕轴突形成髓鞘的现象,以及透射电镜下检测到致密的髓鞘板层结构形成。以上检测结果足以说明体外共培养成髓鞘模型已成功建立。
总之,体外成髓鞘模型是一个有用的实验工具,该模型的建立能帮助我们更好地理解髓鞘形成的内在分子机制。包括髓鞘形成过程中复杂的转录因子调控、髓鞘蛋白的先后表达以及组装髓鞘所必须的髓鞘蛋白的分选等。除此之外,它还对周围神经脱髓鞘疾病的研究和药物的分选提供了方便快捷的工具。
[1]Sherman DL,Brophy PJ.Mechanisms of axon ensheathment and myelin growth[J].Nat Rev Neurosci,2005,6(9):683-690.
[2]Jessen KR,Mirsky R.Negative regulation of myelination:relevance for development,injury,and demyelinating disease[J].Glia,2008,56(14):1552-1565.
[3]Mekhail M,Almazan G,Tabrizian M.Oligodendrocyte-protection and remyelination post-spinal cord injuries:a review[J].Prog Neurobiol,2012,96(3):322-339.
[4]Wei Y,Zhou J,Zheng Z,et al.An improved method for isolating Schwann cells from postnatal rat sciatic nerves[J]. Cell Tissue Res,2009,337(3):361-369.
[5]Garbay B,Heape AM,Sargueil F,et al.Myelin synthesis in the peripheral nervous system[J].Prog Neurobiol,2000,61
(3):267-304.
[6]Päiväläinen S,Nissinen M,Honkanen H,et al.Myelination in mouse dorsal root ganglion/Schwann cell cocultures[J]. Mol Cell Neurosci,2008,37(3):568-578.
[7]Orr DJ,Smith RA.Neuronal maintenance and neurite extension of adult mouse neurones in non-neuronal cell-reduced cultures is dependent on substratum coating[J].J Cell Sci, 1988,91(Pt 4):555-561.
[8]Hall AK.Rodent sensory neuron culture and analysis[J]. Curr Protoc Neurosci,2006,Chapter 3:3-19.
[9]Eshed Y,Feinberg K,Poliak S,et al.Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier[J].Neuron,2005,47(2):215-229.
[10]Court FA,Zambroni D,Pavoni E,et al.MMP2-9 cleavage of dystroglycan alters the size and molecular composition of Schwann cell domains[J].J Neurosci,2011,31(34):12208-12217.
[11]Cosgaya JM,Chan JR,Shooter EM.The neurotrophin receptor p75NTR as a positive modulator of myelination[J]. Science,2002,298(5596):1245-1248.
[12]Court FA,Hewitt JE,Davies K,et al.A laminin-2,dystroglycan,utrophin axis is required for compartmentalization and elongation of myelin segments[J].J Neurosci,2009,29 (12):3908-3919.
[13]Jirsová K,Sodaar P,Mandys V,et al.Cold jet:a method to obtain pure Schwann cell cultures without the need for cytotoxic,apoptosis-inducing drug treatment[J].J Neurosci Methods,1997,78(1/2):133-137.
[14]Vroemen M,Weidner N.Purification of schwann cells by selection of p75 low affinity nerve growth factor receptor expressing cells from adult peripheral nerve[J].J Neurosci Methods,2003,124(2):135-143.
[15]Williams BS,Doyle MD.An Internet Atlas of mouse development[J].Comput Med Imaging Graph,1997,20(6):433-447.
[16]Woodhoo A,Sommer L.Development of the schwann cell lineage:from the neural crest to the myelinated nerve[J]. Glia,2008,56(14):1481-1490.
[17]Désarnaud F,Do Thi AN,Brown AM,et al.Progesterone stimulates the activity of the promoters of peripheral myelin protein-22 and protein zero genes in Schwann cells[J].J Neurochem,1998,71(4):1765-1768.
[18]Melcangi RC,Magnaghi V,Cavarretta I,et al.Age-induced decrease of glycoprotein Po and myelin basic protein gene expression in the rat sciatic nerve.Repair by steroid derivatives[J].Neuroscience,1998,85(2):569-578.
[19]Lewin GR,Barde YA.Physiology of the neurotrophins[J]. Annu Rev Neurosci,1996,19:289-317.
[20]Chan JR,Watkins TA,Cosgaya JM,et al.NGF controls axonal receptivity to myelination by schwann cells or oligodendrocytes[J].Neuron,2004,43(2):183-191.
[21]Eldridge CF,Bunge MB,Bunge RP,et al.Differentiation of axon-related Schwann cells in vitro.I.Ascorbic acid regulates basal lamina assembly and myelin formation[J].J Cell Biol,1987,105(2):1023-1034.
[22]Eldridge CF,Bunge MB,Bunge RP.Differentiation of axon-related Schwann cells in vitro:II.Control of myelin formation by basal lamina[J].J Neurosci,1989,9(2):625-638.
Establishment of a Schwann cell-dorsal root ganglion neuron cocluture model of myelination in the peripheral nervous system
SU Wenfeng,WEI Zhongya,GU Yun,SHEN Yuntian,CHEN Gang
(Key Laboratory of Neuroregeneration,Nantong University,JiangSu 226001)
Objective:To study the mechanism of peripheral nerve myelination,establish a standardized method of Schwann cell and dorsal root ganglion neuron co-culture system of myelination in vitro were established.Methods:SCs were isolated from the sciatic nerves of newborn rat pups(within postnatal 1~3 d).Dorsal root ganglia(DRG)neurons were isolated from the pups of E14~15d SD rats and through the explant culture of DRG.After purification and qualification,these cells were used for co-culture.Then myelin formation were tested by methods of Immune Fluorescent Staining,scanning electron microscopy and transmission electron microscopy.Results:SCs were arranged neatly and their bodies were uniform after purification.The statistical data of immunofluorescence staining showed that the cell purification could reach up to 98% and they could be used for co-culture;DRG neurons were attached to the cell plate steadily,and no hybrid cells could be observed.The immunofluorescence staining of MAG and NF demonstrated that a significant quantity of myelin had formed, and the myelin surrounded the axons tightly;under the scanning electron microscope the process of myelination were obseved while under the transmission electron microscopy showed the typical compact myelin lamellar structure.Conclusions:A stable and reliable model of myelination in vitro has been established and could be used in our subsequent study.
Schwann cells;dorsal root ganglion neurons;myelination;Immune Fluorescent Staining;rat
Q189
A
国家自然科学基金(30800315,31071251);南通大学校级自然科学类科研基金项目(13Z008)。
2014-07-20
1006-2440(2014)04-0310-06
**[作者简介]苏文凤,女,汉族,山东菏泽人,出生于1986年5月,硕士研究生,助理实验员。研究方向:胶质细胞功能研究。 通信作者:陈罡,E-mail:chengang6626@gmail.com
髓鞘是指包绕神经轴突周围的一种层状结构,它既可保护轴突又可加速神经冲动的传导。神经髓鞘化是包括蛋白合成、胞吐、胞吞、细胞骨架运动在内的一个复杂的动态过程,受到神经元和成髓鞘细胞之间的精细调节,涉及到一系列基因和蛋白的变化[1]。髓鞘化异常可导致电信号在神经元间传递障碍,是神经系统炎性脱髓鞘病、脑白质营养不良、阿尔茨海默症、精神分裂症等多种神经系统疾病的病理基础[2]。另外,损伤的神经纤维再生修复、功能的恢复也依赖于轴突的重新髓鞘化[3],因此,研究髓鞘化的机制具有重要的临床意义。体外神经元与施万细胞的共培养成髓鞘模型,一定程度上排除了动物个
体差异、体内其他器官代偿作用和局部微环境变化等体内因素的干扰,是髓鞘化研究的一个有力手段。在体外成髓鞘模型研究中,已有报道表明,由于采用了不同的培养方法导致实验结果在细胞纯度、培养周期、稳定性和可重复性等方面都存在较大的差异[4-8]。本研究参阅大量文献,汲取各方法的优势加以改进,目的是为了建立和推广标准化、简约化的模型构建方法,为开展周围神经髓鞘化的主要调节因素及其作用机制研究提供有效的实验模型。
猜你喜欢
山东医药(2023年23期)2023-09-08 18:33:05
中草药(2022年17期)2022-09-05 05:28:06
交通医学(2021年3期)2021-06-30 17:14:00
解剖学杂志(2021年5期)2021-03-27 12:01:41
遵义医科大学学报(2019年5期)2019-11-29 03:55:24
承德医学院学报(2019年4期)2019-07-09 08:27:08
中国乳品工业(2018年10期)2018-11-16 08:19:24
上海故事(2015年13期)2016-01-22 13:25:09
伴侣(2015年5期)2015-09-10 07:22:44
医学综述(2014年8期)2014-03-08 06:41:48
