
Theoretical study of an energetic material di-1H-1,3,4-triazole derivatives
2014-02-15 06:02HuZHOUZhonglingMAJinlongWANGDongWANG
Defence Technology 2014年4期
Hu ZHOU*,Zhong-ling MAJin-long WANGDong WANG
aNorth University of China,Taiyuan 030051,China
bShanxi Jiangyang Xing'an Explosive Materials Co.Ltd,Taiyuan 030051,China
Theoretical study of an energetic material di-1H-1,3,4-triazole derivatives
Hua ZHOUa,*,Zhong-liang MAa,Jian-long WANGa,Dong WANGb
aNorth University of China,Taiyuan 030051,China
bShanxi Jiangyang Xing'an Explosive Materials Co.Ltd,Taiyuan 030051,China
Computations by density functional theory(DFT)method are performed on a series of di-1H-1,3,4-triazole derivatives with different substituents and linkages.The heat of formation(HOF)is predicted by the designed isodesmic reactions.The predicted results reveal that-N3and -N=N-groups are effective structural units for increasing the HOF values of the di-1H-1,3,4-triazole derivatives.The HOMO-LUMO gap is affected by the substituents and linkage groups.Detonation performance is evaluated using the Kamlet-Jacobs approach based on the calculated density and HOF.The results indicate that-NO2,-NF2,-NH-,-NH-NH-and-N=N-groups are helpful for enhancing the detonation properties of di-1H-1,3,4-triazole derivatives.The bond dissociation energy and bond order of the weakest bonds are analyzed to investigate their stability.It is observed that the-CH2-,-CH2-CH2-and-CH=CH-groups are effective structural units for improving the stabilities of these derivatives.Considering the detonation performance and the stability,fve compounds are screened as the potential candidates for high energy density materials.
Di-1H-1,3,4-triazole;Density functional theory;Heat of formation;Detonation velocity;Detonation pressure
1.Introduction
The synthesis and theoretical study on the nitrogen-rich compounds have received considerable interest in recent years[1-8].Energetic nitrogen-rich compounds are potential candidates for high energy density materials(HEDMs)due to their large number of N-N and N-C bonds in molecular skeleton,which leads to high density,high positive heat of formation,good oxygen balance,and good stability.Triazole-based energetic materials are most prominent among the nitrogen-rich compounds which appear to be a better compromise with high energy,performance and high stability due to nitrogen catenation and aromaticity[9].Among them, 1H-1,3,4-triazole is an effective structural unit.
It is well known that properties of high energy,performance and high stability are often improved by making structural modifcations.The formation of molecular complexes(e.g., di-,tri,and polymers)is a conceivable way to increase the density and thermostability and improve the material properties of propellants and explosives[10].Therefore,as we know, triazole connected on the N atom of heterocyclic is more prone to substitution reaction.Hence,it is easily to synthesize the high nitrogen compounds with better comprehensive properties.Thermodynamic calculations conducted at Los Alamos National Laboratory identifed 2,5,2′,5′-tetranitro-1,1′-bi-1,3,4-triazole(TNBT)as target molecule was likely to exhibit high performance[11].2,5,2′-triazido-1,1′-azo-1,3,4-triazole was synthesized[12],which shows that tetrazene(N-N= N-N)can improve HOF but reduce the stability.However,the systematic and comprehensive molecular design still lacks for di-1H-1,3,4-triazole-based high energy density materials.
In this paper,the HOF,electronic structures,energetic properties,and thermal stabilities of a series of di-1,3,4-triazole derivatives with various substituents(-NH2,-NO2, -NF2,-N3)and different linkages(-,-CH2-,-NH-, -CH2-CH2-,-NH-NH-,-CH=CH-,-N=N-)were systematically studied based on the density functional theory (DFT)method.
2.Computational methods
The object molecules,the series of 1H-1,3,4-triazole derivatives of which the molecules are numbered as(A1-A5, B1-B5,C1-C5,D1-D5,E1-E5,F1-F5 and G1-G5),are classifed into seven groups,as shown in Fig.1.The hybrid DFT-B3LYP method[13,14]in combination with 6-31G(d,p)basis set is used for structure optimization,which has been proved[15-18]to give quite reliable energies,molecular structures,and other properties.All quantum mechanical calculations were performed with Gaussian 03 program package [19].The optimized structure of each molecule corresponds to only one local energy minimum on the potential energy surface without imaginary frequency.
The isodesmic reaction method is employed to calculateHOF.Here we design an isodesmic reaction in which the numbers of all kinds of bonds are kept constant to decrease the calculation errors ofHOF.Because of the electronic circumstances of reactants and products are very similar in the isodesmic reactions,the errors of electronic correction energies can be counteracted,therefore,the errors of calculatedHOFcan be greatly reduced[20].In these designed reactions,the basic structural unit of 1,3,4-triazole skeleton is kept constant, and the complex molecules are split into sample molecules. This method had been shown to be reliable[3,15,21-23].
TheHOFof di-1H-1,3,4-triazole derivatives at 298 K was obtained by isodesmic reactions,which is given as follows
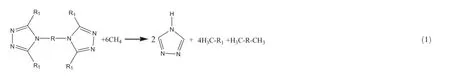
whereR1=-H,-NH2,-NO2,-NF2,-N3.R=-,-CH2-,
-NH-,-CH2CH2-,-NH-NH-,-CH=CH-,-N=N-. For the isodesmic reaction,the heat of reaction ΔH298at 298 K can be calculated from the following equation
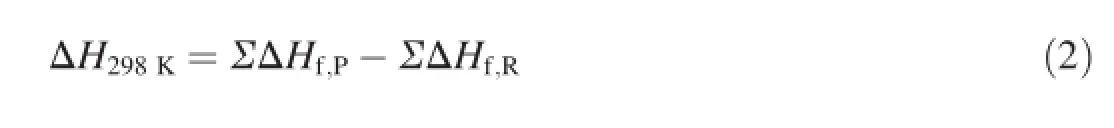
where ΔHf,Pand ΔHf,Rare theHOFs of products and reactants at 298 K,respectively.Since the experimentalHOFs of 1H-1,3,4-triazole,CH3NF2,CH3N3,and CH3N=NCH3are unavailable,the additional calculations were carried out.TheHOFof CH3NF2was calculated by the replacement reaction CH3NH2+F2→CH3NF2+H2using G2 theory[17,23].TheHOFvalues of 1H-1,3,4-triazole,CH3N3and CH3N=NCH3were obtained at G2 level from the atomization reaction CaHbOcNd→aC(g)+bH(g)+cO(g)+dN(g).The experimentalHOFsofreferencecompoundsCH4,CH3NH2, CH3NO2,CH3NHCH3,CH3CH3,CH3CH2CH3,CH3CH2CH2CH3,CH3NHNHCH3and CH3CH=CHCH3are available [24-26].
TheHOFs of 1H-1,3,4-triazole-based derivatives can be estimated when the heat of reaction ΔH298Kis known. Therefore,the primary thing is to calculate ΔH298K.ΔH298Kcan be calculated by using the following expression
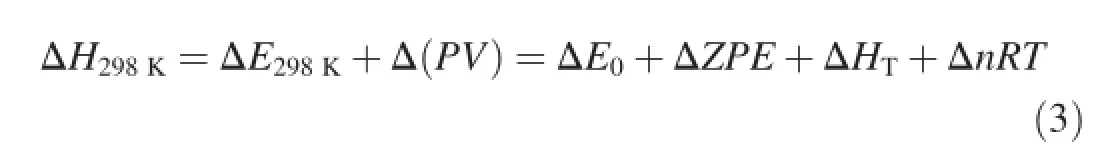
where ΔE0is the change in total energy between the products and the reactants at 0 K;ΔZPEis the difference between the zero-point energies(ZPE)of the products and the reactants; and ΔHTis the thermal correction from 0 to 298 K.Δ(PV) equals ΔnRTfor the reaction of ideal gas.For the isodesmic reaction here,Δn=0,so Δ(PV)=0.
Since the condensed phases of most energetic compounds are solid,the solid-phase heat of formation(ΔHf,solid)is required in order to calculate the detonation properties.According to Hess's law of constant heat summation[27],the gas-phase heat of formation(ΔHf,gas)and heat of sublimation (ΔHsub)can be used to evaluate the solid-phase heat of formation

Politzer et al.[28-30]pointed out that the heat of sublimation of energetic compounds correlates well with the molecular surface area and the electrostatic interaction indexThe empirical expression for this approach is shown below
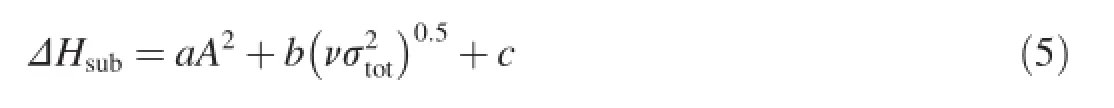
whereAis the surface area of the 0.001 electrons/bohr3isosurface of the electronic density of the molecule;ν describes the degree of balance between positive and negative potentials on the isosurface;andis a measure of the variability of the electrostatic potential on the molecular surface.The coeffcientsa,b,andcwere determined by Rice et al.:a=2.670×10-4kcal mol-1A-4,b=1.650 kcal mol-1,andc=2.966 kcal mol-1[31].The descriptorsA,ν,andwere calculated using the computational procedures described by Felipe et al.[32].This approach has been demonstrated to be a reliable way to predict the heats of sublimation of many energetic compounds[31-33].
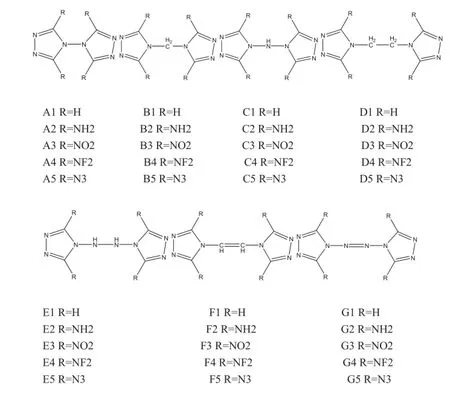
Fig.1.Molecular frameworks of di-1H-1,3,4-triazole derivatives.
The empirical Kamlet-Jacobs equations[34,35]were employed to estimate the values ofDandPfor high energy density materials.The equations are as follows
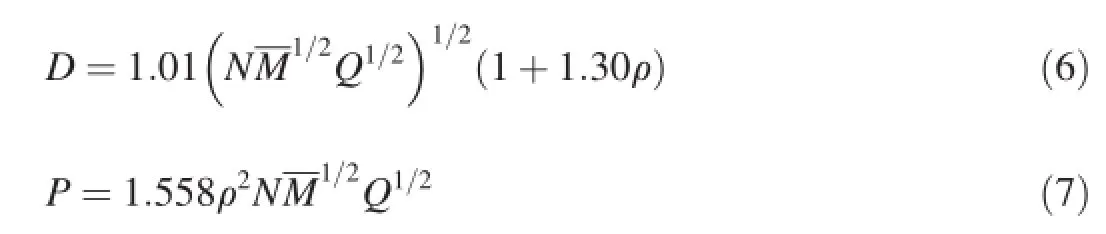
whereDis the detonation velocity(km/s);Pis the detonation pressure(GPa);Nis the moles of detonation gas per gram of explosive;Mis the average molecular weight of these gases;Qis the heat of detonation(Cal/g);and ρ is the density of explosives(g/cm3).For known explosives,Qand ρ can be measured experimentally;thus,DandPcan be calculated according to Eqs.(6)and(7).However,for some compounds,Qand ρ cannot be estimated from experimental measurements.Therefore,Qand ρ need frst to be calculated to estimateDandP.
The heat of detonationQwas estimated by the HOF difference between the products and explosives according to the principle of exothermic reactions,i.e.,all the N atoms are combined into N2,F atoms form HF with H atoms,or are combined into F2without H atoms,and oxygen atoms form H2O before CO2.If the content of O is not suffcient to satisfy full oxidation of H and C atoms,the remaining H atoms will convert into H2,and C atoms will exist as solid-state C.In the Kamlet-Jacobs equations,the detonation products are assumed to be CO2(or C),H2O(or H2or HF or F2),and N2,so the energy released in the decomposition reaction is maximized.The correspondingDandPvalues can be estimated using the values of density andQ.The theoretical molecular density used in this work was slightly greater than the practical loaded density.Therefore,according to the Kamlet-Jacobs equations,the values ofDandPcan be regarded as their upper limits.
For each di-1H-1,3,4-triazole derivative,the theoretical density was obtained from the molecular weight divided by the average molecular volume.The volume is defned as that inside the density contour of 0.001 electrons/bohr3,which was estimated using Monte Carlo integration.We performed 100 single-point calculations for each optimized structure to get the average volume at B3LYP/6-31G(d,p)level.The crystal density can be improved by introducing the interaction index[36]:
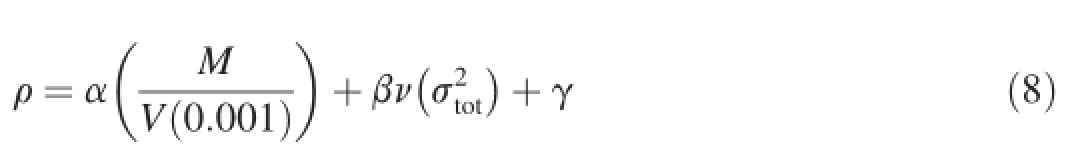
whereMis the molecular mass(g/mol);andV(0.001)is the volume of the 0.001 electrons/bohr3contour of electronicdensity of the molecule(cm3/molecular).The coeffcients α, β,and γ are 0.9183,0.0028,and 0.0443,respectively[36].
The HOMO-LUMO gap between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO)can be correlated with the sensitivity of the molecules[37].The HOMO-LUMO gap of di-1H-1,3,4-triazole derivatives was predicted using DFT at B3LYP/ 6-31G(d,p)level.
The bond strength can be estimated by bond dissociation energy,which is fundamental to understand the chemical processes[38].The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction AB(g)-A(g)+B(g),which is the bond dissociation enthalpy of the molecule A-B by defnition[39].For many organic molecules,the terms“bond dissociation energy”and“bond dissociation enthalpy”often appear interchangeably in the Ref.[40].
Thus,at 0 K,the homolytic bond dissociation energy can be given in terms of
BDE0(A-B)=E0(A·)+E0(B·)-E0(A-B) (9)
The bond dissociation energy(BDE)with zero-point energy(ZPE)correction can be calculated by the following equation
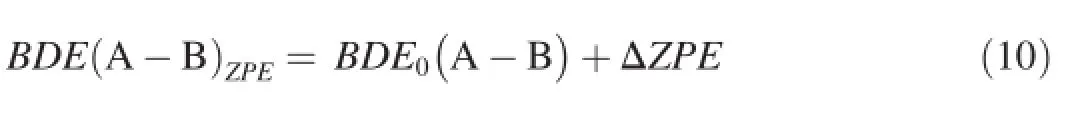
where ΔZPEis the difference between theZPEs of the products and the reactants without basis set superposition error (BSSE).
3.Results and discussion
3.1.Heat of formation
Here we investigated the effects of different substituents and linkages on the heats of formation of di-1H-1,3,4-triazole derivatives.Table 1 lists the total energy,ZPE,thermal correction and relative error for 13 reference compounds in the isodesmic reactions.The experimentalHOFsofCH4, CH3NH2,CH3NO2,CH3CH3,CH3NHCH3,CH3CH2CH3, CH3(CH2)2CH3,CH3(NH)2CH3,CH3CH=CHCH3and CH3N=NCH3were taken from Refs.[24-26].To validate the reliability of our algorithm,the HOF values of these compounds were calculated at G2 level from the atomization reaction.The results show that the HOF values were in accordance with the experimental values.The largest relative error for these compounds is 4.32%.Therefore theHOFvalues from the G2 level are expected to be reliable in this work.
Table 2 summarizes the total energy,ZPE,thermal correction,andHOFfor the di-1H-1,3,4-triazole derivatives. All the derivatives exhibited high positiveHOF.It is clear that, when-N3and-NO2were attached to the parent compound, theHOFvalues increased compared with the unsubstituted case,whereas the opposite was true for the substituents-NH2and-NF2.In addition,it is worth noticing that-N3groupextremely enhances theHOFvalue compared with parent di-1H-1,3,4-triazole.These observations indicate that-N3group plays an essential role in increasing theHOFvalues of di-1H-1,3,4-triazole derivatives.
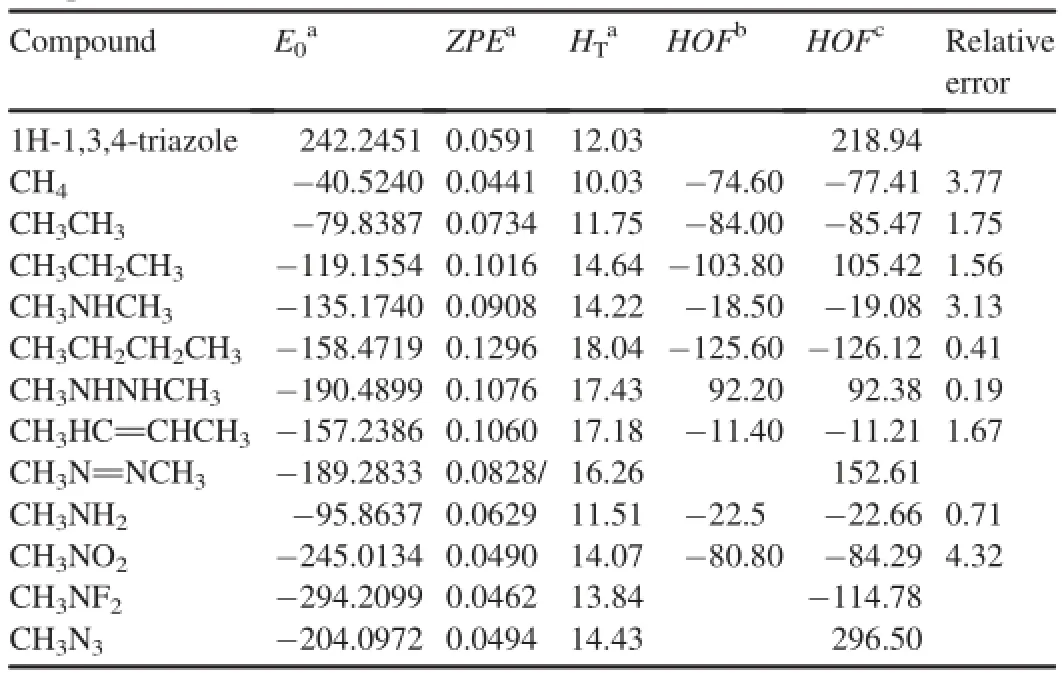
Table 1Calculated total energies(E0),zero-point energies(ZPE),thermal corrections (HT),relative errors(%)and heats of formation(HOFs)for the reference compounds.
Fig.2 shows a comparison of theHOFvalues of di-1H-1,3,4-triazole derivatives with different linkages.TheHOFvalues of the structures having-NH-linkage(C series), -NH-NH-linkage(E series)and-N=N-linkage(G series)were higher than those of the directly linked di-1H-1,3,4-triazoles(A series)with the same substituents.But when the linkages were-CH2-(B series),-CH2-CH2-(D series)or -CH=CH-(F series),the situations were opposite.And the order ofHOFvalue of the structures possessing linkage groups can be given as-N=N->-NH-NH->-NH->->-CH=CH->-CH2->-CH2-CH2-.We noticed that the di-1H-1,3,4-triazole with the conjugated linkage-N=N-and-CH=CH-had higherHOFvalues than the corresponding ones with the unconjugated linkage-NH-NH-and -CH2-CH2-.The reason for this situation may be that the two 1H-1,3,4-triazoles and the conjugated linkage forms a big conjugated system.Di-1H-1,3,4-triazoles linked by the azo linkage(-N=N-)had the largestHOFvalues among the derivatives with the same substituents.This indicates that azo (-N=N-)is an effective linkage for increasing theHOFvalues of di-1H-1,3,4-triazole derivatives.TheHOFvalue of G5(di-1H-1,3,4-triazole with linkage group of-N=N-and substituent of-N3)is the largest one among these derivatives, which is in agreement with the analysis result.
3.2.Electronic structure
The HOMO-LUMO gap is correlated with the sensitivity of material within the limitations ofDFT[42].In general,the smaller the HOMO-LUMO gap is,the easier the electron transition is,and the larger the sensitivity is.Table 3 lists theNote:E0andZPEare in a.u.;HTandHOFare in kJ/mol.The scaling factor is 0.98 forZPEand 0.96 forHT.[41]. highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO)energies and the energy gaps(ΔELUMO-HOMO)for the di-1H-1,3,4-triazole derivatives.It is found that the HOMO-LUMO gaps of all substituted derivatives are smaller than those of the corresponding unsubstituted di-1H-1,3,4-triazoles.It is interesting to note that HOMO-LUMO gaps are in the order of -H>-NH2>-NF2>-N3>-NO2except for G series. While the order is-H>-NF2>-NO2>-N3>-NH2in G series,the substituents have the effect on the HOMO-LUMO gapsofvariousseries.The orderwasfound to be ->-CH2-CH2->-CH2>-NH>-NH-NH->-CH=CH->-N=N-by comparing the derivatives with the same substituents and different bridges.Among these derivatives,A1 had the largest HOMO-LUMO gap,whereas G5 had the smallest one.Since the molecule with smaller HOMO-LUMO gap was expected to have a higher reactivity and a lower stability in the chemical or photochemical processes with electron transfer or leap[43,44],it might be inferred that the-N=N-bridged di-1H-1,3,4-triazole had the highest reactivity among these bridged derivatives.
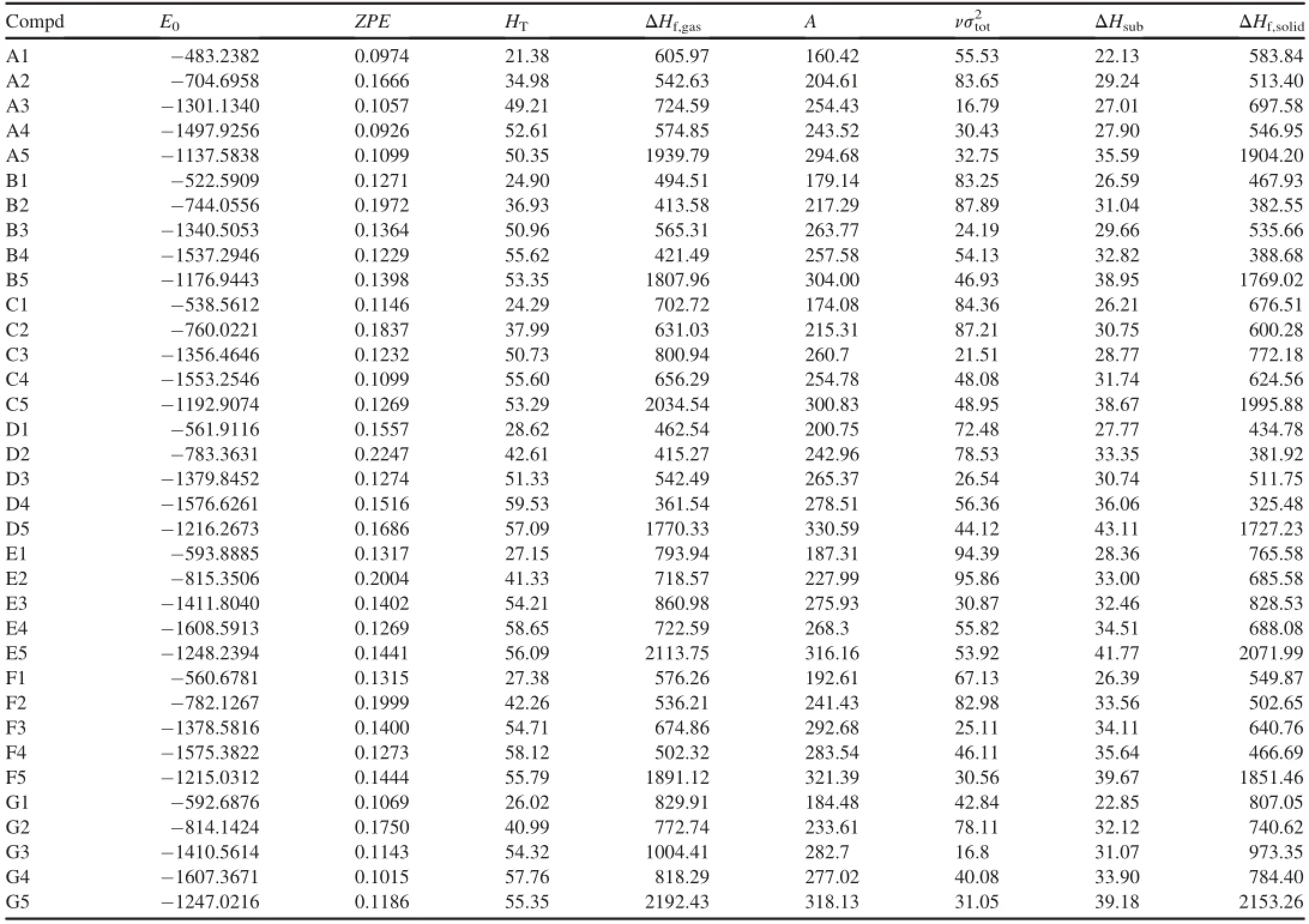
Table 2Calculated total energies(E0),zero-point energies(ZPE),thermal corrections(HT),molecular properties,heats of sublimation(ΔHsub),and heats of formation (ΔHf,gas,ΔHf,solid)for di-1H-1,3,4-triazole derivatives.

Fig.2.Comparison ofHOFs of di-1H-1,3,4-triazoles with different substituents and linkages.
3.3.Energetic properties
Detonation velocity(D),detonation pressure(P)and heat of detonation(Q)are important performance parameters for anenergetic material.The Kamlet-Jacobs equations show that the density ρ is a key factor to infuenceDandP[45].Thus ρ is an important physical property for all energetic materials. Table 4 lists the calculated ρ,Q,D,Pand oxygen balance (OB)of the di-1H-1,3,4-triazole derivatives.For a comparison, the experimental detonation properties of 1,3,5-trinitro-1,3,5-triazinane (RDX) and 1,3,5,7-tetranitro-1,3,5-tetrazocane (HMX)are also listed in Table 4.
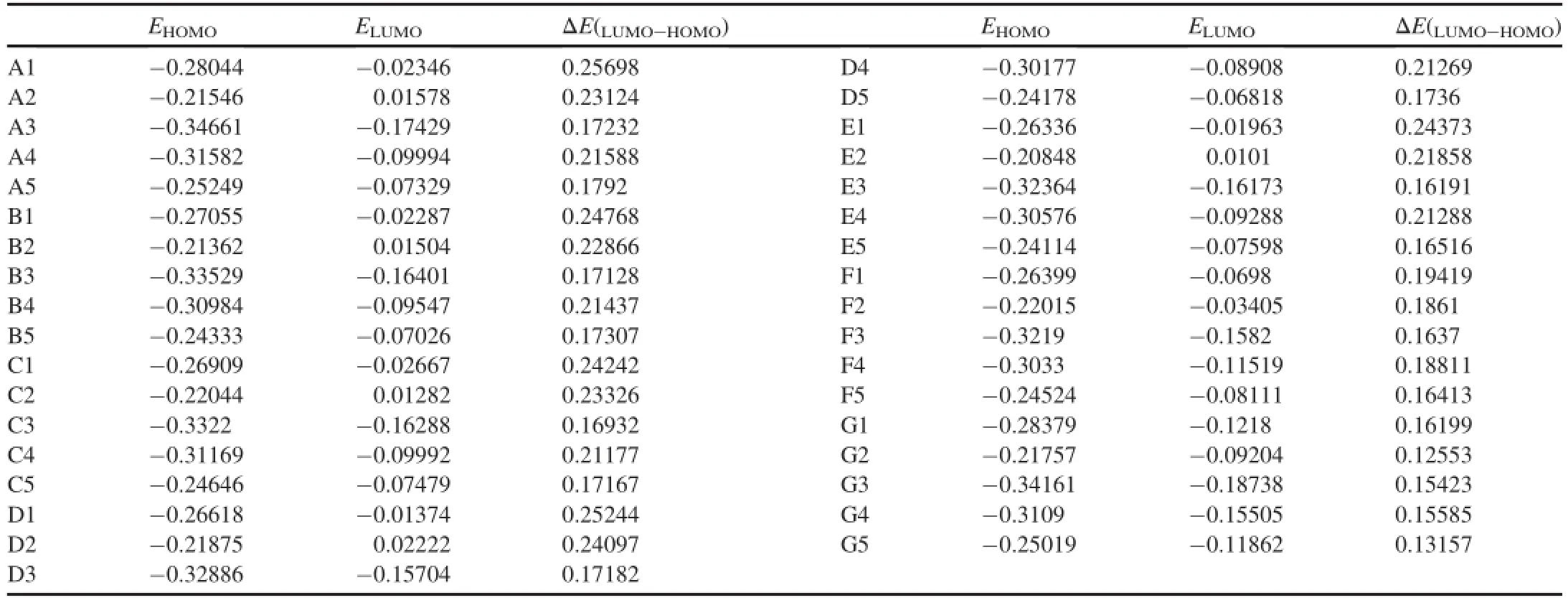
Table 3Calculated HOMO and LUMO energies(a.u.)and energy gaps ΔE(LUMO-HOMO)of di-1H-1,3,4-triazole derivatives.
The density values of all the substituted di-1H-1,3,4-triazole are larger than those of unsubstituted molecules.The -NF2group increases the density of the di-1H-1,3,4-triazole derivatives compared with other substituents.To improve the density,the order can be given as-NF2> -NO2>-N3>-NH2.When the linkages are-CH2-CH2-(D series) and-CH=CH-(F series),the density values of the di-1H-1,3,4-triazole derivatives are smaller than those of the directly linked di-1H-1,3,4-triazoles with the same substituents.When the linkages are-CH2-(B series),-NH-(C series), -NH-NH-(E series)and-N=N-(G series),the density of di-1H-1,3,4-triazole with-N3or without substituent is higher than that of the corresponding directly linked di-1H-1,3,4-triazole.This indicates that the effects of the linkages -CH2-,-NH-,-NH-NH-and-N=N-on the density values were coupled to those of the substituents.Specially,the -NH-linkage with-NF2substituent(C4)has the largest ρ values among all of the derivatives.
The calculated heat of detonation in Table 4 shows that the substituents-NO2,-NF2,and-N3increase the heat of detonation compared with that of the corresponding unsubstitued di-1H-1,3,4-triazoles,whereas the opposite is true for -NH2.The different linkage groups also have different effects on the detonation heats of the di-1H-1,3,4-triazole derivatives. On the whole,-N=N-is an effective group for enhancing the detonation heats of di-1H-1,3,4-triazole derivatives.
When the substituent is-NH2,the density values of derivatives increase but the heat of formation decrease,soDandPvalues of A2,F2 and G2 are larger than those of their unsubstituted homolog in-NH2derivatives.When-NO2, -NF2and-N3are attached to di-1H-1,3,4-triazole,they can increase both density and heat of formation,leading to largerDandPvalues of all the substituted derivatives compared with their unsubstituted homolog.
It is also found thatDandPvalues of-NH-, -NH-NH-,and-N=N-derivatives are higher than those of the directly linked di-1H-1,3,4-triazoles with the same substituents.But-CH2-,-CH2-CH2-and-CH=CH-linkage groups are unhelpful in increasing the detonation properties of the di-1H-1,3,4-triazole derivatives.Based on the above analyses,it can be concluded that-NO2,-NF2, -NH-,-NH-NH-and-N=N-groups are the effective structural units to enhance the detonation properties of di-1H-1,3,4-triazole derivatives.
Fig.3 shows the calculatedDandPvalues of the di-1H-1,3,4-triazole derivatives together with commonly used explosives RDX and HMX.It is found A3,A4,B4,C3,C4,D4, E3,E4,F4,G3 and G4 exhibit better detonation performances (DandP)compared with HMX,which is one of the most widely used energetic ingredients of various high-performance explosives and propellant formulations.Although it was reported that A3(TNBT)and G5 have been successfully synthesized[46,47],some of their detonation properties have still undetermined yet.In addition,the syntheses of other energetic compounds have not yet been reported.If A4,B4,C3,C4,D4, E3,E4,F4,G3 and G4 could be synthesized,they would have higher detonation performance.Thus,we should make further investigations on this kind of compounds in the future.
3.4.Stability
The bond dissociation energy(BDE)provides useful information for understanding the stability of a molecule. Generally speaking,the lower the bond dissociation energy is, the weaker the bond would be,which will be the trigger bond. The sensitivity and stability of the energetic materials aredirectly relevant to the bond strength,which is commonly measured by bond dissociation energy(BDE).Therefore, based on the Mulliken bond order values,we selected relatively weak bond in the same molecule to calculate BDE,and then screened out the trigger bond based on BDE values.Table 5 lists the bond dissociation energy(BDE)and the Mulliken bond order(BO)of relatively weaker bonds of di-1H-1,3,4-triazole derivatives.It is found that some bonds have higher BDE but lower bond order in a same molecular,for example, B3,B4,C3 and C4.Thus,the stability of the di-1H-1,3,4-triazole derivatives should not be judged only by the bond order criterion,but also based onBDE.
From Table 5 it can be known that the BDE value of the linkage bond is smaller than those of other bonds in the same molecule except for B3,D3,D4,F3 and F4.It may be inferredthat the linkage bond may be the weakest one and the breakage of linkage is possible in thermal decomposition,and theBDEvalues are affected by both substituent and linkage group.It is found by comparing the derivatives with the same substituents and different linkages that-CH2-,-CH2-CH2- and -CH=CH-linkage groups can increase theBDEvalues substantially.For the same linkage group but different sunstituent,all the substituent groups are attached to the parent compounds,theirBDEvalues decrease,and the order can be given as-H>-NO2-NF2>-N3>-NH2.
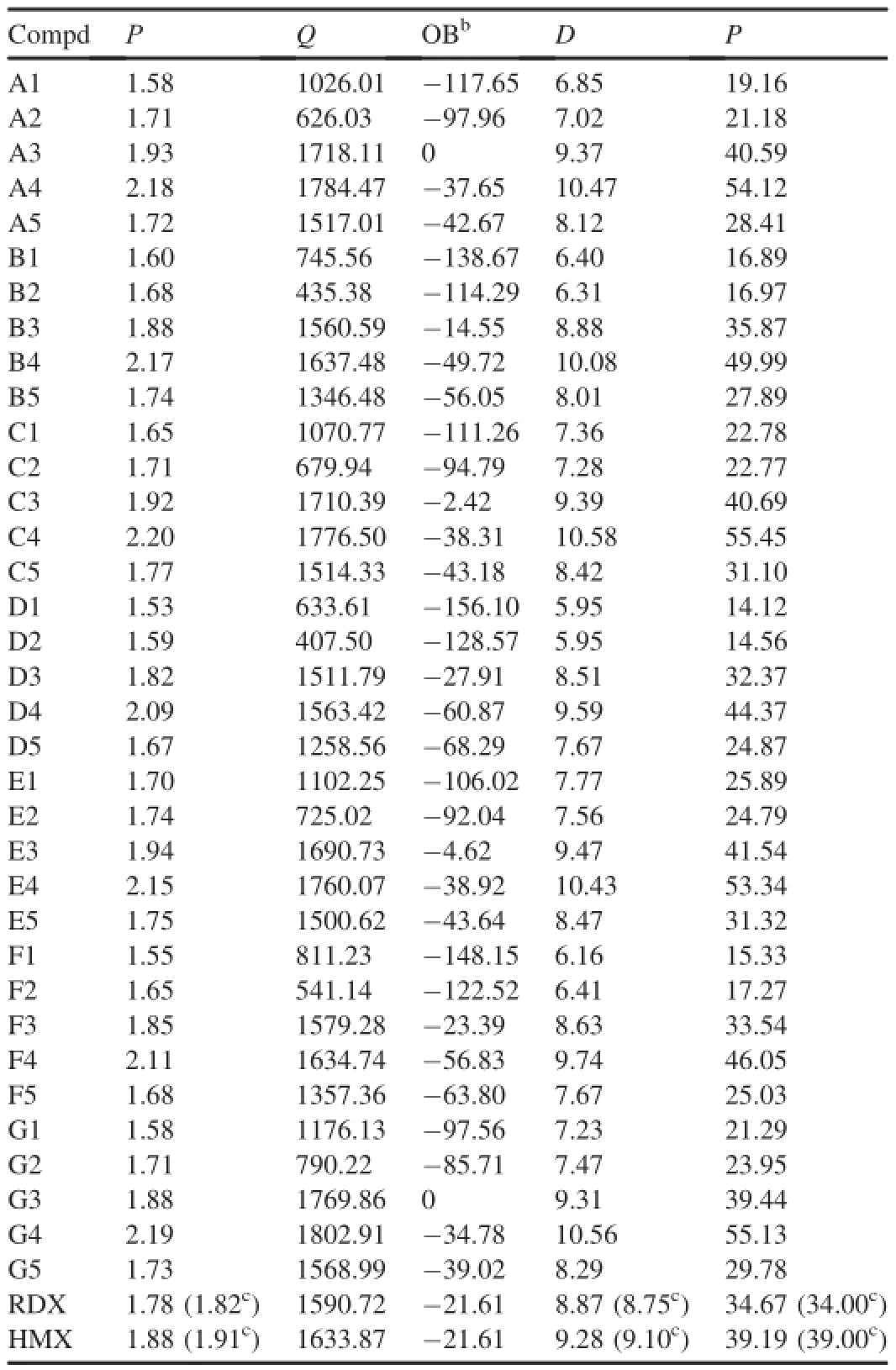
Table 4Predicted densities(ρ,g/cm3),heats of detonation(Q,Cal/g),detonation velocities(D,km/s),detonation pressures(P,GPa),and oxygen balance(OB)for di-1H-1,3,4-triazole derivatives.a

Fig.3.Detonation velocity and pressure of di-1H-1,3,4-triazole derivatives.
Fig.4 showsBDEof the weakest bonds of the di-1H-1,3,4-triazole derivatives along with RDX and HMX.It is easily noticedthattheBDEvalueoftheweakestbondinthederivatives ishigherthanthatofHMXwhenthelinkagegroupsare-CH2-, -CH2-CH2-and-CH=CH-.As it is well known,a good nitrogen-rich HEDM candidate not only has excellent detonation properties but also exist stably.A3,B4,D4,E3 and F4 possessbetterdetonationperformances(DandP)andstabilities (BDE)compared with HMX.Therefore,these fve compounds could be considered as potential candidates for HEDM with enhanced performance and reduced sensitivity.
4.Conclusions
In this paper,the DFT-B3LYP method was used to study the heat of formation(HOF),electronic structure,energetic properties,and stability of a series of di-1H-1,3,4-triazole derivatives with different linkages and substituents.The results show that-N3and-N=N-groups play a very important role in increasing the HOFs of di-1H-1,3,4-triazole derivatives.A1 has the largest HOMO-LUMO gap,whereas G5 has the smallest one.The effects of substituents on the HOMO-LUMO gap combine with those linkage groups.
The calculated detonation velocities and detonation pressures of the di-1H-1,3,4-triazole derivatives indicate that -NO2,-NF2,-NH-,-NH-NH-and-N=N-groups are the effective structural units for enhancing the detonation properties.An analysis of the bond dissociation energy(BDE)for several relatively weak bonds suggests that-CH2, -CH2-CH2-,and-CH=CH-groups are the effective structural units for improving the stability of di-1H-1,3,4-triazole derivatives.A3,B4,D4,E3 and F4 were screened as potential candidates for HEDM with enhanced performance and reduced sensitivity.
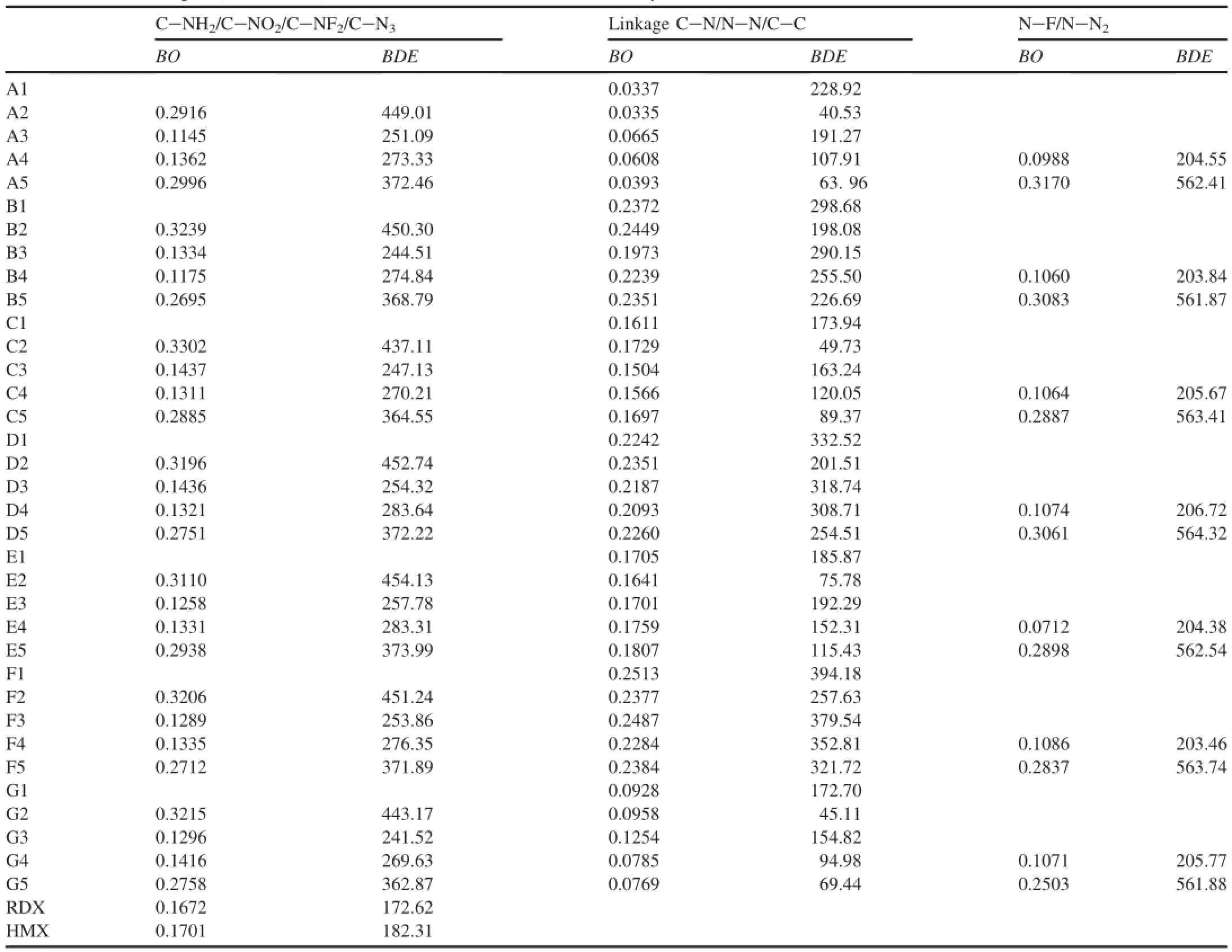
Table 5Bond dissociation energies(BDE,kJ/mol)and Mulliken bond orders of the relatively weak bonds in di-1H-1,3,4-triazole derivatives.
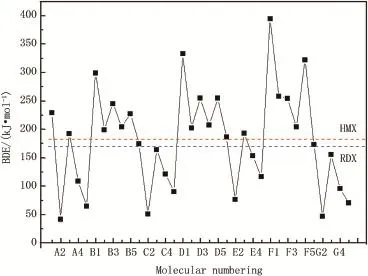
Fig.4.Bond dissociation energies of the weakest bonds for di-1H-1,3,4-triazole derivatives.
[1]Xu XJ,Xiao HM,Ju XH,Gong XD,Zhu WH.Computational studies on polynitrohexaazaadmantanes as potential high energy density materials.J Phys Chem A 2006;110(17):5929-33.
[2]Gutowski KE,Rogers RD,Dixon DA.Accurate thermochemical properties for energetic materials applications.I.Heats of formation of nitrogen-containing heterocycles and energetic precursor molecules from electronic structure theory.J Phys Chem A 2006;110(42):11890-7.
[3]Wei T,Zhu WH,Zhang XW,Li YF,Xiao HM.Molecular design of 1,2,4,5-tetrazine-based high-energy density materials.J Phys Chem A 2009;113(33):9404-12.
[4]Wei T,Zhu WH,Zhang J,Xiao HM.DFT study on energetic tetrazolo-[1,5-b]-1,2,4,5-tetrazine and 1,2,4-triazolo-[4,3-b]-1,2,4,5-tetrazine derivatives.J Hazard Mater 2010;179(1-3):581-90.
[5]Zhang CC,Zhu WH,Xiao HM.Density functional theory studies of energetic nitrogen-rich derivatives carbon-bridged diiminotetrazoles. J Theor Comput Chem 2011;967(2-3):257-64.
[6]Joo YH,Shreeve JM.Energetic mono-or bis(oxy)-5-nitroiminotetrazoles. Angew Chem Int Ed 2010;49(40):7320-3.
[7]Pan Y,Zhu WH,Xiao HM.Design and selection of nitrogen-rich bridged di-1,3,5-triazine derivatives with high energy and reduced sensitivity. J Mol Model 2012;18(7):3125-38.
[8]Qui L,Xiao HM,Gong XD,Ju XH,Zhu WH.Theoretical studies on the structures,thermodynamic properties,detonation properties,and pyrolysis mechanisms of spiro nitramines. J Phys Chem A 2006;110(10):3797-807.
[9]Ghule Vikas D.Computational studies on the triazole-based high energy materials.J Comput Theor Chem 2012;992(4):92-6.
[10]Korkin AA,Bartlett RJ.Theoretical prediction of 2,4,6-trinitro-1,3,5-triazine(TNTA).A new,powerful,high energy density material.J Am Chem Soc 1996;118(48):12244-5.
[11]Coburn MD,Harris BW,Hayden HH,Lee K-Y,Stinecipher MM, Hayden HH.Explosives synthesis at Los Alamos.Ind Eng Chem Prod Res Dev 1986;25(1):68-72.
[12]Li SH,Shi HG,Sun CH,Li XT,Pang SP,Yu YZ,Zhao XQ.Synthesis and crystal structyre of nitrogen-rich compound:2,5,2′-triazido-1,1′-azo-1,3,4-triazole.J Chem Crystallogr 2009;39(2):13-6.
[13]Lee C,Yang W,Parr RG.Development of the Colle-Salvetti correlationenergy formula into a functional of the electron density.Phys Rev B 1988;37(2):785-9.
[14]Becke AD.Density-functional thermochemistry.III.The role of exact exchange.J Chem Phys 1993;98(7):5648-52.
[15]Chen ZX,Xiao JM,Xiao HM,Chiu YN.Studies on heats of formation for tetrazole derivatives with density functional theory B3LYP method.J Phys Chem A 1999;103(40):8062-6.
[16]Selmi M,Tomasi J.Ab-initio calculations and semiempirical corrections to obtain enthalpies of formation of hydrocarbons through isodesmic reactions.J Phys Chem 1995;99(16):5894-8.
[17]Ju XH,Li YM,Xiao HM.Theoretical studies on the heats of formation and the interactions among the difuoroamino groups in polydifuoroaminocubanes.J Phys Chem A 2005;109(5):934-8.
[18]Jursic BS.Computing the heat of formation for cubane and tetrahedrane with density functional theory and complete basis set ab initio methods. J Mol Struct 2000;499(1):137-40.
[19]Frisch MJ,Trucks GW,Schlegel HB,et al.Gaussian 03,revision B.03. Pittsburgh,PA:Gaussian Inc.;2003.
[20]Hahre WJ,Radom L,Schleyer PVR,Pole JA.Ab initio molecular orbital theory.New York:Wiley;1986.p.386-90.
[21]Zhang XW,Zhu WH,Xiao HM.Comparative theoretical-studies of energetic substituted carbon and nitrogen-bridged difurazans.J Phys Chem A 2010;114(1):603-12.
[22]Chen PC,Chieh YC,Tzeng SC.Density functional calculation of the heats of formation for various aromatic nitro compounds.J Mol Struct 2003;634(1):215-24.
[23]Ju XH,Wang X,Bei FL.Substituent effects on heats of formation,group interactions,and detonation properties of polyazidocubanes.J Comput Chem 2005;26(12):1263-9.
[24]Dean JA.Lange's handbook of chemistry.15th ed.New York:McGraw-Hill;1999.p.82-8.chapter 6.
[25]David RL.Handbook of chemistry and physics.84th ed.Boca Raton: CRC Press;2003-2004.
[26]Afeefy HY,Liebman JF,Stein SE.Neutral thermochemical data.In: Linstrom PJ,Mallard WG,editors.NIST Chemistry WeBook:NIST standard reference database number 69;2000.National Institute of Standards and Technology,Gaithersburg.Respectively.
[27]Atkins PW.Physical chemistry.Oxford:Oxford University Press;1982.
[28]Politzer P,Lane P,Murray JS.Computational characterization of a potential energetic compound:1,3,5,7-tetranitro-2,4,6,8-tetraazacubane. Cent Eur J Energ Mater 2011;8(1):39-52.
[29]Politzer P,Murray JS.Some perspectives on estimating detonation propertiesofC,H,N,O compounds.CentEurJEnerg Mat 2011;8(3):209-20.
[30]Politzer P,Murray JS,Grice ME,Desalvo M,Miller E.Calculation of heats of sublimation and solid phase heats of formation.Mol Phys 1997;91(5):923-8.
[31]Byrd EFC,Rice BM.Improved prediction of heats of formation of energetic materials using quantum chemical calculations.J Phys Chem A 2006;110(1):1005-13.
[32]Bulat FA,Toro-Labbe A,Brinck T,Murray JS,Politzer P.Quantitative analysis of molecular surfaces:areas,volumes,electrostatic potentials and average local ionization energies. J Mol Model 2010;16(11):1679-91.
[33]Jaidann M,Roy S,About-Rachid H,Lussier LS.A DFT theoretical study of heats of formation and detonation properties of nitrogen-rich explosives.J Hazard Mater 2010;176(1-3):165-73.
[34]Kamlet MJ,Jacobs SJ.Chemistry of detonations.I.Simple method for calculating detonation properties of carbon-hydrogen-nitrogen-oxygen explosives.J Chem Phys 1968;48(3):23-35.
[35]Kamlet MJ,Ablard JE.Chemistry of detonations.II.Buffered equilibrium.J Chem Phys 1968;48(2):36-40.
[36]Politzer P,Martinez J,Murray JS,Concha MC,Toro-Labbe A.An electrostatic interaction correction for improves crystal density prediction.Mol Phys 2009;107(19):2095-101.
[37]Badders NR,Wei C,Aldeeb AA,Rogers WJ,Mannan MS.Predicting the impact sensitivities of polynitro compounds using quantum chemical descriptor.J Energ Mater 2006;24(1):17-33.
[38]Benson SW.Thermochemical kinetics.2nd ed.New York:Wiley Interscience;1976.
[39]Mills I,Cvitas T,Homann K,Kallay N,Kuchitsu K.Quantities,units,and symbols in physical chemistry.1st ed.Oxford:Blackwell Science;1988.
[40]Blanksby SJ,Ellison GB.Bond dissociation energies of organic molecules.Acc Chem Res 2003;36(4):255-63.
[41]Scott AP,Radom L.Harmonic vibrational frequencies:an evaluation of Hartree-Fock,Møller-Plesset,quadratic confguration interaction, density functional theory,and semiempirical scale factors.J Phys Chem 1996;100(41):16502-13.
[42]XU XJ,Zhu WH,Xiao HM.DFT studies on the four polymorphs of crystalline CL-20 and the infuences of hydrostatic pressure on epsilon-CL-20 crystal.J Phys Chem B 2007;111(8):2090-7.
[43]Dong HS,Zhou FF.High energy explosives and correlative physical properties.Beijing:Science Press;1989.
[44]Hoffmanx R.Symmetry requirements for stabilization of one class of diradicals.J Chem Soc D 1969;112(5):240-1.
[45]Talawar MB,Sivabalan R,Mukundan T,Muthurajan H,SIkder AK, Gandhe BR,Rao AS.Environmentally compatible next generation green energetic materials(GEMs).J Hazard Mater 2009;161(2-3):589-607.
[46]Coburn MD,Harris BW,Hayden HH,Lee K-Y,Stinecipher MM.Explosives synthesis at Los Alamos.Ind Eng Chem Prod Res Dev 1986;25(1):68-72.
[47]Li SH,Pang SP,Li XT,Yu YZ,Zhao XQ.Synthesis and crystal structure of novel highly nitrogen-containing compound of polyazidotriazole.Chin J Org Chem 2008;28(4):727-31.
Received 25 April 2014;revised 14 August 2014;accepted 15 August 2014 Available online 5 October 2014
*Corresponding author.
E-mail address:zhouhua654312@163.com(H.ZHOU).
Peer review under responsibility of China Ordnance Society.
http://dx.doi.org/10.1016/j.dt.2014.08.002
2214-9147/Copyright©2014,China Ordnance Society.Production and hosting by Elsevier B.V.All rights reserved.
Copyright©2014,China Ordnance Society.Production and hosting by Elsevier B.V.All rights reserved.

- Defence Technology的其它文章
- Analysis of parameter estimation using the sampling-type algorithm of discrete fractional Fourier transform
- Nitrogen analogs of TEX-A computational study
- Neural network modeling to evaluate the dynamic fow stress of high strength armor steels under high strain rate compression
- Scale-up synthesis and characterization of 2,6-diamino-3,5-dinitropyrazine-1-oxide
- Experiment and simulation of launching process of a small-diameter steel cartridge case
- Reliability sensitivity analysis based on multi-hyperplane combination method