
分子动力学方法模拟Be高温和高压的状态方程和力学性质
2014-02-13 02:31黄晓玉何朝政宋海珍殷复荣于荣梅
西南民族大学学报(自然科学版) 2014年6期
黄晓玉, 何朝政, 宋海珍, 殷复荣, 于荣梅
(南阳师范学院物理与电子工程学院, 河南 南阳 473061)
分子动力学方法模拟Be高温和高压的状态方程和力学性质
黄晓玉, 何朝政, 宋海珍, 殷复荣, 于荣梅
(南阳师范学院物理与电子工程学院, 河南 南阳 473061)
利用分子动力学方法, 采用修正镶嵌原子势函数, 在两组势参数下模拟Be单晶的等温、等压状态方程.通过径向分布函数, 均方根位移随时间的变化分析Be的结构信息.比较两组参数的模拟结果, 第一组势参数作用下体系原子间作用较小, 较易压缩.两组势参数300K等温压率比较,第二组势参数的模拟结果与实验符合的更好.对于弹性常数,第二组势参数不能考虑温度和压强的影响.用第一组势参数计算Be单晶的弹性常数, 根据弹性常数随压强的变化判断材料的低温相变, 相变压强为320GPa, 与第一性原理计算符合较好; 弹性常数随温度增加而减小, 与实验结果相符.总体而言, 第一组势函数能更全面的反映Be单晶的结构特点.
分子动力学方法; 修正型嵌入原子势; 状态方程; 弹性常数
金属铍(Be)是一种重要的战略材料, 在武器系统、原子能、航空航天等工业领域中有着很广泛的应用[1]. 虽然Be原子的电子排布(1s22s2)非常简单, 但是由Be 原子组成的金属却具有非常独特的物理性质如Debye温度高、熔点高、熔化潜热大、热中子吸收截面小、泊松比小等.Be金属薄层被用作激光聚变靶丸的烧蚀层, 另外, 金属Be还被用作核反应堆的中子减速剂、反射体材料和中子源等, 这些都需要在高温高压的条件下进行, 高温和高压下金属Be结构和性质的研究吸引众多研究者的注意.
近年来人们主要研究了金属Be的相变以及弹性性质[2-4].在室温和一个大气压下金属Be是六角密排结构(hcp), 研究发现无论是改变金属Be所处的压强还是温度条件都会使金属Be的结构由hcp转变为体心立方结构(bcc).常压(保持一个大气压下)下, 改变温度发现金属Be在达到熔化之前大约30K时就由hcp相转变为bcc相[5-6].在实验方面, 使用金刚石压砧(DAC) 压缩金属Be, 通过观察电阻的变化来测量金属Be相变的压强, 一直到40 GPa时, Reichlin等人[7]还是一直没有观察到相变, 金属Be仍然是 hcp 结构.进一步提高实验手段, 采用大功率的X 射线衍射方法观测, 还是采用DAC压缩金属Be直到压强达到171GPa, Nakano 等人[8]还是没有观察到晶体Be结构的变化.目前更高压强下的实验数据还没有找到.在理论方面, Benedict等人[9]利用第一性原理方法, 发现金属Be在400GPa附近时才发生相变, 并做出了Be的hcp相、bcc相和液相的三相图, 但是目前实验上还得不到验证.Fen Luo等人[10]用第一性原理方法计算高温、高压下金属Be的热力学性质, 给出相变压强为390Gpa, 其计算结果中高压下的弹性常数C44较实验数据偏大.Aimin Hao等人[11]用第一性原理方法计算高压下金属Be的结构稳定性和弹性常数, 给出相变压强为320Gpa.对与弹性常数, Marie等人[12]利用超声方法研究金属Be的弹性模量在300K到1000K之间随温度的变化趋势, 数据和原有实验有明显的差异.K.Kádas等人[2]利用第一性原理计算各弹性常数, 体积弹性模量随温度的变化.总之, 关于金属Be相变的压强, 第一性原理相关研究结果还存在较大的争议.另外, 金属Be弹性模量随温度的变化与实验结果差别较大.
目前, 对于金属Be的高温高压下的热力学性质的理论研究还不是很详尽.因此, 我们需要采用其他方法和模型来进行研究.本文采用分子动力学的方法和和两组修正型嵌入原子势(Modified embedded atom method (MEAM))参数计算高温、高压下金属Be的状态方程和弹性常数, 将计算数据与实验和第一性原理的结果进行比较, 并给出相关分析.
1 计算方法
分子动力学方法是基于牛顿力学基本原理, 通过对运动方程积分得到系统粒子的位臵构型和动量, 并运用统计力学方法得到物质的宏观性质.本文模拟体系是晶格结构为hcp结构, 晶格常数a为2.27Å, c为3.59Å, 含有2048个Be原子的块金属固体.初始速度是按300K温度下麦克斯韦波尔兹曼分布提取的, 每次模拟的结果都是在系统达到平衡之后进行统计平均的.模拟体系选取的时间步长是1fs, 选择三维周期性边界条件, 选用的系综包括等温等压系综(NPT)、和正则系综(NVT)等.
分子动力学(MD)方法是利用原子间相互作用势来计算作用在原子上的力.因此, 相互作用势就是模拟最关键的输入要素.本文中, 采用Baskes提出的hcp结构金属的MEAM相互作用势[13].由Daw和Baskes提出的EAM势, 能够很好的描述金属间的相互作用[14-15].他们从准原子理论出发, 把系统中的每一个原子均看作是嵌在由其它原子形成的主晶格中的杂质.运用密度泛函理论并考虑局域密度近似得到系统的总能量为:


我们有必要对本文用到的两组势参数给出说明和解释.其中, 第二组势参数是1993年M I Baskes等人[13]以HCP结构金属为研究对象开发的多体势, 这组势参数只有在温度为0K的情况下才能很好的模拟材料的力学、热力学性质, 即使在很小的温度下Be的晶体结构就会变得很不稳定.第一组势参数是2007年V.V.Dremov等人[16]在MEAM理论框架下, 对势参数进行新的优化得到的.优化后的参数能够很好的模拟材料的力学, 热力学性质.下面我们用表格的形式分别给出两组势参数:
表1 金属Be hcp相的MEAM参数其中)是结合能,是平衡时的最近邻距离,是能量函数的指数衰退因子,是嵌入函数的屏蔽因子,是电子密度的指数衰退因子,t(h)是电子密度的权重因子.Table 1 Parameters of the MEAM potential for beryllium(,rc(Å)equilibrium nearest-neighbor distance,the exponential decay factor for the universal energy function,A the scaling factor for the embedding function,the exponential decay factor for the atom densities,t(h)the weighting factors )

表1 金属Be hcp相的MEAM参数其中)是结合能,是平衡时的最近邻距离,是能量函数的指数衰退因子,是嵌入函数的屏蔽因子,是电子密度的指数衰退因子,t(h)是电子密度的权重因子.Table 1 Parameters of the MEAM potential for beryllium(,rc(Å)equilibrium nearest-neighbor distance,the exponential decay factor for the universal energy function,A the scaling factor for the embedding function,the exponential decay factor for the atom densities,t(h)the weighting factors )

2 结果和讨论
结果与讨论部分由三个部分组成: 1)比较两组势参数下的高温、高压状态方程, 了解两组势参数的特点和区别; 2)比较两组势参数下的径向分布函数, 均方位移随时间的变化, 了解两组势参数的特点和区别; 3.)用第一组势参数计算体系高温、高压下的弹性常数, 体积弹性模量, 将结果与对应的第一性原理计算、实验结果比较.
2.1 体系的状态方程
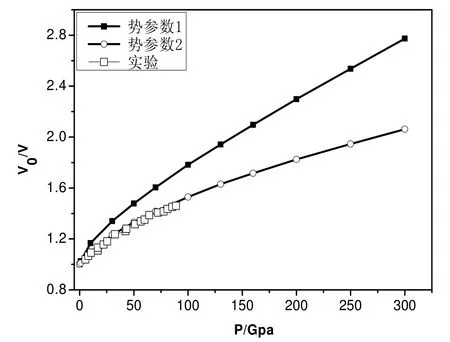
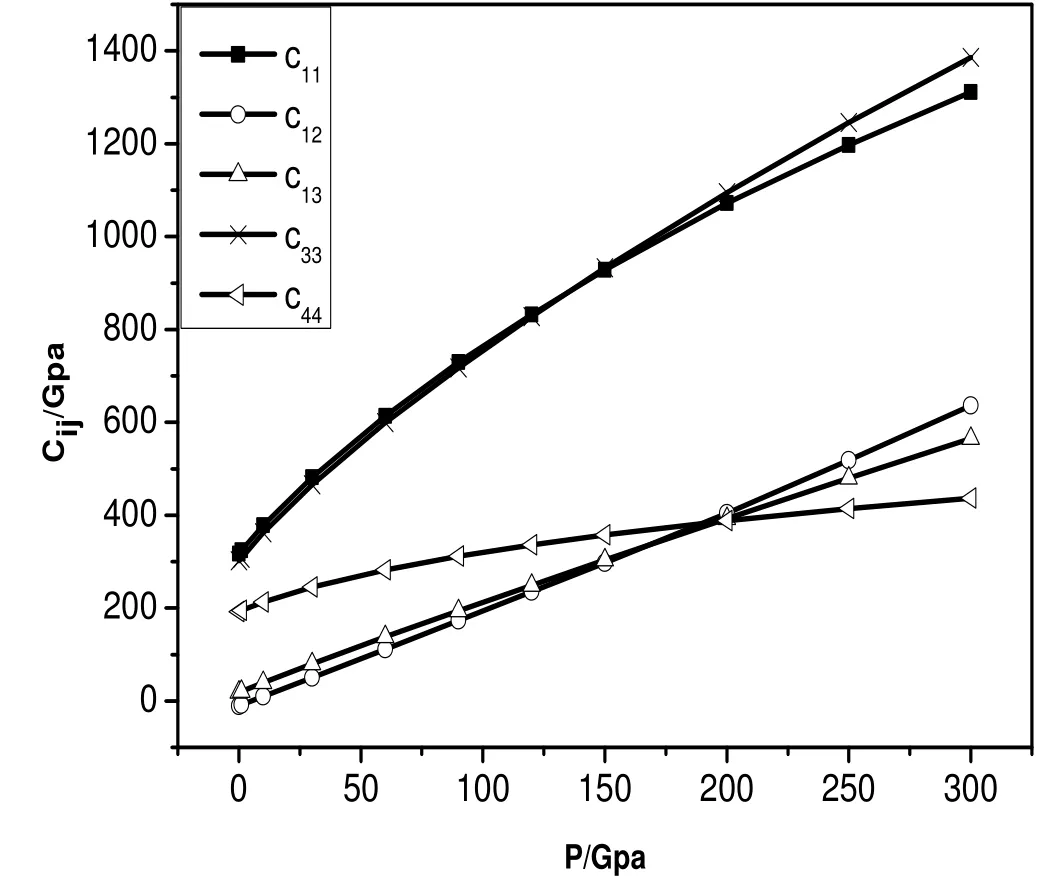
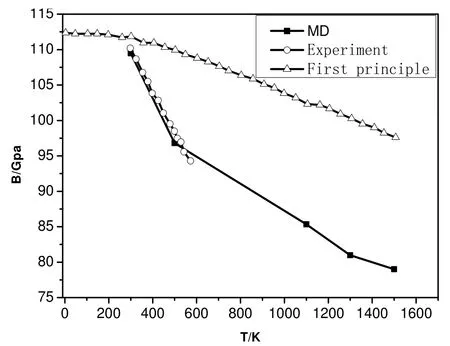
首先, 利用两组势函数模拟体系的等温状态方程曲线(恒温300K), 如图1所示: 在第一组势函数作用下, 平衡后体系的体积V1, 相应的第二组势函数作用下体系的体积V2.压强小于150Gpa时:V1>V2,压强增加至150Gpa时:V1=V2, 压强大于150Gpa时, V1 图1 两组势参数300K等温状态方程比较Fig.1 Isothermal equation of state at 300K with two potential parameters 图2 两组势参数300K等温压率比较 Fig.2 Isothermal compressibility at 300K with two potential parameters. 2.2 体系的结构分析 在统计力学中, 径向分布函数(rdf)指的是给定某个粒子的坐标, 其他粒子在空间的分布几率.径向分布函数可以用来研究物质的有序性, 能够很好的描述非晶和晶体材料的原子结构特征.如图4所示: 温度为300K、压强为0.1Mpa时, 两种势函数作用下体系的径向分布函数.在第一组势函数作用下, 三个峰的位臵分别对应第一、二、三最近邻距离: 2.25Å,4.15Å,6.15Å, 第二组势参数相应的值分别为: 2.25Å,3.75Å,5.65Å.可以看出第一组势函数作用下, 体系长程有序结构更松弛. 分子动力学计算系统中的原子由起始位臵不停移动, 每一瞬间各原子的位臵皆不相同.粒子位移平方的平均值称为均方位移(MSD).如图5a,b)所示: 体系在第一组势函数作用下,常压、温度分别为300K和1500K时均方根位移随时间的变化.300K时, MSD随时间增加变化不大, 1500K时MSD随时间增加线性增长; 如图6a,b)所示: 体系在第二组势函数作用下常压、温度分别为300K和1500K时均方根位移随时间的变化.同样, 300K时, MSD随时间增加变化不大, 1500K时MSD随时间增加线性增长.这说明1500K时, Be晶体的原子结构已趋于无序, 基本熔化, 与已知的熔点1550K基本一致[18].两图的区别在于: 第一组势函数作用下体系的MSD数值较大; 1500K时第一组势函数作用下MSD随时间增加线性增长的斜率较大, 易膨胀. 图3 两种势函数等压状态方程比较Fig.3 Isobaric equation of state with two potential parameters 图4 两种势函数径向分布函数比较 Fig.4 Radial distribution function at 300K with two potential parameters 图5 势参数1下体系的均方位移Fig.5 Mean square displacement with the first potential parameters 图6 势参数2下体系的均方位移 Fig.6 Mean square displacement with the second potential parameters 表2 两种势函数下等压状态方程分段拟合Table 2 Curve fitting of isobaric equation of state 2.3 体系的弹性常数 材料的弹性常数描述了它对所加外力的响应, 或者可以这样说, 弹性常数描述了为维持一个给定的形变所需的应力.对体系施加不同的变形, 根据总能量和应变的关系, 拟合弹性常数的具体数值.其中第二组势参数的计算结果有大的无规则波动, 不能有效考虑温度和压强的效应.因此, 将第一组势参数模拟的弹性常数和相应的实验数据、第一性计算数据比较, 给出分析结果. 图7 MD模拟各向异性弹性常数值Fig.7 Anisotropic elastic constants by MD simulation 图8 MD模拟单轴弹性模量 Fig.8 Uniaxial elastic modulus by MD simulation 图9 体积弹性模量随压强变化(MD与第一性原理比较)Fig.9 The variation of Bulk modulus with pressure (MD simulation and first principles study ) 图10 MD体积弹性模量值与实验值、第一性计算值比较 Fig.10 The variation of Bulk modulus with temperature (experiment and first principles study ) 对于HCP结构的Be晶体, 它的独立的弹性常数为: C11, C12, C13, C33和C44.如图7所示: 压强范围为0~300Gpa各弹性常数随压强的变化.在指定压强范围内, 各弹性常数随压强的变化线性增加.其中, C11和C33数值接近, 变化趋势很接近, 随压强增加速度最快; C44随压强增加速度最慢; C12和C13数值接近, 变化趋势也很接近, 随压强增加速度居中.不同弹性常数的增加并不是完全等比例的, 这说明, Be单晶的弹性各向异性关系在不同压强下也不一样.HCP结构的Be晶体的单轴弹性常数分别为: Ca, Cb, Cc.Ca=0.5*(C11-C12), Cb=C44, Cc=1/6*(C11+2*C33+C12-4*C13), 如图8所示: 压强范围为0~300Gpa, 单轴弹性常数随压强的变化.其中, Cb,Cc的数值随压强增加速度较快; Cc的数值随压强增加速度较慢; 压强在150Gpa~300Gpa范围, Ca的数值随压强增加变化趋于平缓, 数值变化很小. 如前所述, Be单晶的常温高压相变行为: hcp~bcc, 相变压强的确定尚存争议.本文通过Be单晶体积弹性模量随压强的变化给出分子动力学方法对于相变的判断.如图9所示, 给出压强范围为0~400Gpa, MD方法和第一性原理计算方法, Be单晶的体积弹性模量随压强的变化.在0~300Gpa范围内, MD模拟结果与Hao A等人的计算结果符合的很好, 和Luo F的数据有较小的差别. 压强为320Gpa时, 体积弹性模量发生突变, 可以推测Be单晶发生相变行为.这一结果和Hao A等人给出的相变压强一致, 和Luo F等人给的结果有一定偏差.如图10所示: 三种方法体积弹性模量随温度变化, 弹性常数随温度的增加而减小.MD计算结果与实验数据符合的较好[2],第一性原理方法计算数据与实验数据相比有较大偏差. 本研究利用两组势函数模拟Be单晶的等温、等压状态方程; 径向分布函数和均方根位移; 弹性常数、体积弹性模量.可以得出结论: 第一组势函数作用下, 体系的可压缩性强, 原子间的作用力较弱; 长程有序结构更加松弛; 均方位移数值较大且随时间的变化率值较大. 两组势参数300K等温压率比较,第二组势参数的模拟结果与实验符合的更好.但是, 第二组势函数计算弹性常数时, 不能有效地考虑压强和温度的效应.第一组势函数模拟的弹性常数随压强的变化显示体系的各项异性特点, 其中C轴可压缩性较强; 体积弹性模量在压强为320Gpa时有突变, 据此可推测Be单晶hcp到bcc结构的相变行为, 这一结果和已有的第一性原理计算符合较好.体积弹性模量随温度增加而减小, 与已有的实验数据符合较好, 弥补第一性原理数据的偏差.本文将两组势函数的模拟结果比较分析, 研究结果说明: 第一组势函数能够更全面的反映Be单晶的结构特点.准确的势函数是分子动力学模拟的关键要素, 希望我们的研究结果对后来的理论模拟工作提供参考. [1]张友寿, 秦有钧, 吴东周, 等.铍的粉末冶金工艺及焊接研究进展[J].焊接学报, 2001, 22: 6-10. [2]KÁDAS K, VITOS L, AHUJA R, JOHANSSON B, et al.Temperature-dependent elastic properties of α-beryllium from first principles[J].Phys Rev B, 2007, 76: 235109. [3]ROBERT G, LEGRAND P, BERNARD S.Multiphase equation of state and elastic moduli of solid beryllium from first principles[J].Phys Rev B, 2010, 82:104118. [4]宋海峰, 刘海风.金属铍热力学性质的理论研究[J].物理学报, 2007, 56:2833-2838. [5]MARTIN A J, MOORE A.1959 J.Less-Common Met.1 85. [6]FRANCOIS M M.contre, Proc Int Conf Grenoble.Presses Univ de France, Paris, 1965. [7]REICHLIN R L.Measuring the electrical resistance of metals to 40 GPa in the diamond-anvil cell[J].Rev Sci Instrum, 1983, 54: 1647-1652 [8]NAKANO K, AKAHAMA Y, KAWAMURA H.X-ray diffraction study of Be to megabar pressure[J].J Phys: Condens Matter, 2002, 14: 10569-10575. [9]BENEDICT L X, OGITEU T, TRAVE A, et al.Calculations of high pressure properties of beryllium: Construction of a multiphase equation of state[J].Phys Rev B, 2009, 79: 064106-064111. [10]LUO F, CAI L C, CHEN X R, JING F Q, et al.Ab initio calculation of lattice dynamics and thermodynamic properties of beryllium[J].J Appl Phys, 2012, 111: 053503-053508. [11]HAO A, ZHU Y.First-principle investigations of structural stability of beryllium under high pressure [J].J Appl Phys, 2012, 112: 023519-023524. [12]NADAL M H,BOURGEOIS L.Elastic moduli of beryllium versus temperature: Experimental data updating [J].J Appl Phys, 2010, 108: 033512-033517. [13]BASKES M I, JOHNSON R A.Modified embedded atom potentials for HCP metals[J].Modelling Simul Mater Sci Eng, 1994, 2: 147-152. [14]DAW M S, BASKES M I.Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals[J].Phys Rev B, 1984, 29: 6443-6448. [15]DAW M S, FOILES S M, BASKES M I.The embedded-atom method: a review of theory and applications[J].Mater Sci Rep, 1993, 9: 251-256. [16]DREMOV V V, KARAVAEV A V, Kutepov A L, et al.Molecular Dynamics Simulation of Thermodynamic and Mechanical Properties of Be and Mg[J].AIP Conf Proc, 2007, 955: 305-310. [17]VELISAVLIEVIC N, CHESNUT G N, VOHRA Y K.Structural and electrical properties of beryllium metal to 66 GPa studied using designer diamond anvils[J].Phys Rev B, 2002, 65: 172107-172112. Study of equations of state and mechanical property of Be by molecular dynamics method HUANG Xiao-yu, HE Chao-zheng, SONG Hai-zhen, YIN Fu-rong, YU Rong-mei Beryllium is an important strategic nuclear material, which was used in weapons system, atomic energy, aerospace fields. Based on these applying requirements, study of beryllium on structure and properties was necessary. This paper uses molecular dynamics method and modified embedded atom potential to study equations of state of beryllium under high temperature and high pressure with two sets of potential parameters. Structure information of beryllium was analyzed by the radial distribution function and mean square displacement. To compare the computer simulation results with two sets of potential parameters, with the first potential parameters, forces between atoms were smaller, more compressible. For the elastic constants, the second potential parameters can not consider the effects of temperature and pressure; elastic constants changed with temperature and pressure were computed with the first potential parameters. The results were in good accordance with the experiments and the first principles calculations. There is a sudden change when the elastic constant changed with the pressure which can be guessed as the phase transition. The pressure was 320Gpa which was in accordance with the the first principles calculations. In short, the first potential parameters can reflect the structural characteristics of beryllium more comprehensive than the first one. molecular dynamics method; modified embedded atom potential; equation of state; elastic constant O52 A 1003-4271(2014)06-0921-07 10.3969/j.issn.1003-4271.2014.06.21 2014-10-08 黄晓玉(1985-), 女, 汉族, 河南南阳人, 博士, 讲师, 研究方向: 材料状态方程、热力学性质, E-mail: huangxiaoyu198510@163.com. 国家自然科学基金青年科学基金(批准号: 11304167); 河南省教师教育改革重点项目(批准号: 2013-JSJYZD-053); 南阳师范学院科研基金(批准号: ZX2013020、ZX2014088).







3 结论
( School of Physics and Electronic Engineering, Nanyang Normal University, Nanyang 473061, P.R.C.)
猜你喜欢
数学理论与应用(2022年1期)2022-04-15
煤气与热力(2021年6期)2021-07-28
宜春学院学报(2020年9期)2020-12-03
原子与分子物理学报(2020年5期)2020-03-17
四川文理学院学报(2020年5期)2020-02-12
图学学报(2019年1期)2019-03-02
北京航空航天大学学报(2017年1期)2017-11-24
浙江大学学报(理学版)(2017年3期)2017-05-18
燕山大学学报(2015年4期)2015-12-25
原子与分子物理学报(2015年3期)2015-11-24
