
4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′1″]三联苯的合成
2014-02-02 08:45高嫒嫒赵群星闫晓亮
液晶与显示 2014年4期
高嫒嫒,赵群星,陈 龙,李 涛,闫晓亮
(西安彩晶光电科技股份有限公司,陕西 西安 710065)
1 引 言
随着液晶显示技术的不断发展,液晶显示面板的世界市场在进一步扩大,已经广泛地应用在电子显示产品上,如电视、计算机屏幕、笔记型计算机、移动电话或个人数字助理等。大屏幕显示是目前显示市场上的普遍需求,随着大屏幕显示技术的发展,对彩色液晶材料也提出了更高的要求。VA-TFT模式和IPS模式由于其高对比度、宽视角以及快速响应等特点,是最具发展前景的LCD技术。在液晶材料发展趋势中,针对LCD技术的变化,近几年焦点都放在响应速度的提高上:要提高响应速度,液晶的黏度需要下降,或者通过液晶盒变薄来作改善,但却会使其色彩鲜艳度受到影响。为了更好地改善液晶电视在动画显示方面的质量,除了通过改善显示技术外,开发新型低粘度的液晶材料也是一条捷径。
端烯类液晶单体与同结构烷基末端单体相比,具有黏度小、熔点低、清亮点高、低温稳定好的优点,因而由它们调制的混合液晶具有黏度低、黏度随温度变化率低和低温稳定性好的特点,目前大量使用在TFT-LCD混晶中[1-3]。丁烯类液晶是新型端烯类液晶的发展新方向,丁烯类的新型低黏度的液晶材料是改善液晶电视活动画面显示质量的重要途径。目前,丁烯类液晶是高档STN液晶和TFT混晶中一个重要组份,能显著提高混晶的折射率各向差异性。目前丁烯类液晶的合成开发报道虽然不多[4-9],但是该类产品是TFT-LCD高档混晶中的一个主要成分,对改进混晶的性能有着显著作用,是一个具有广阔前景的液晶单体。
本文拟合成化合物:4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′1″]三联苯,是丁烯端烯类液晶中的一个代表化合物。
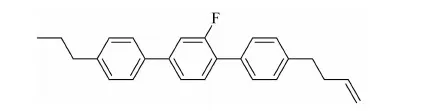
与目前液晶配方中常用的乙烯端烯类液晶相比,该化合物的分子构型的线性增加,有效改变了单体分子的长宽比,使液晶相区进一步拓宽,黏度降低、熔点降低、清亮点提高、低温稳定性更好,并且还在三联苯上引入侧向氟原子,氟原子较高的电负性将影响到分子的偶极矩,使液晶分子具有低黏度,适中的介电各向异性、高电阻率和高电荷保持率等特点。
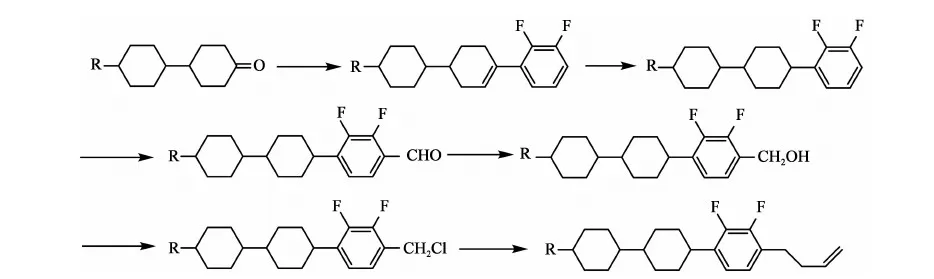
文献报道合成丁烯端基的方法主要有2种:
方法一[5]:
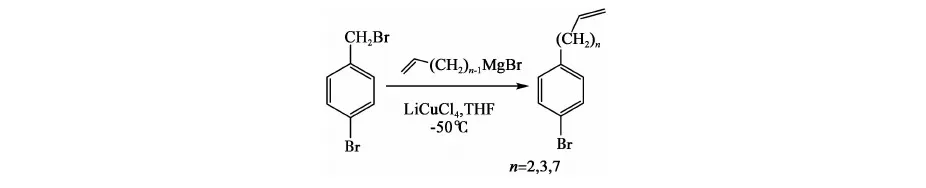
方法二[10]:
参考上述文献及以往合成经验,本文设计合成路线如下:
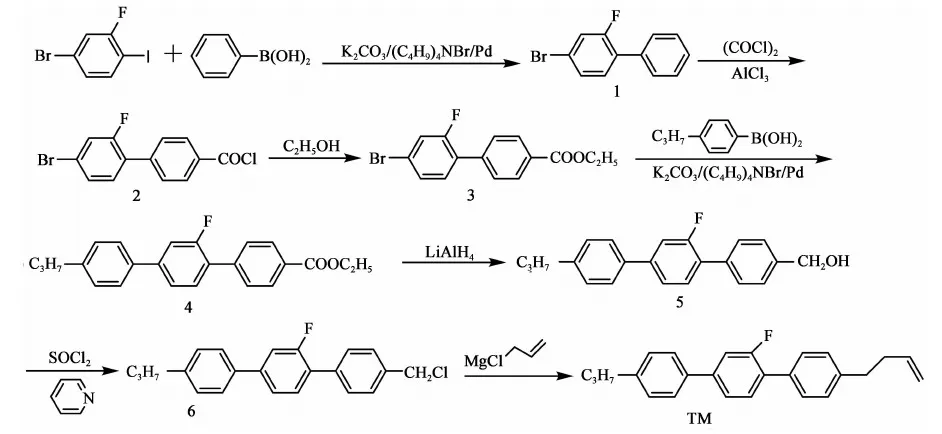
此法以2-氟-4-溴碘苯和苯硼酸为原料,经过对接、酰化、酯化反应得到2-氟-4-溴-1-乙酸乙酯基联苯(化合物3),再与丙基苯硼酸对接、还原、氯代得到4-氯甲基-2′-氟-4″丙基-[1,1′,4′1″]三联苯(化合物6),再与3-氯丙烯的格氏试剂对接,共七步反应得到目标产物,总收率(以2-氟-4-溴碘苯计)21.7%。
2 实 验
2.1 原材料
原材料规格要求见表1。
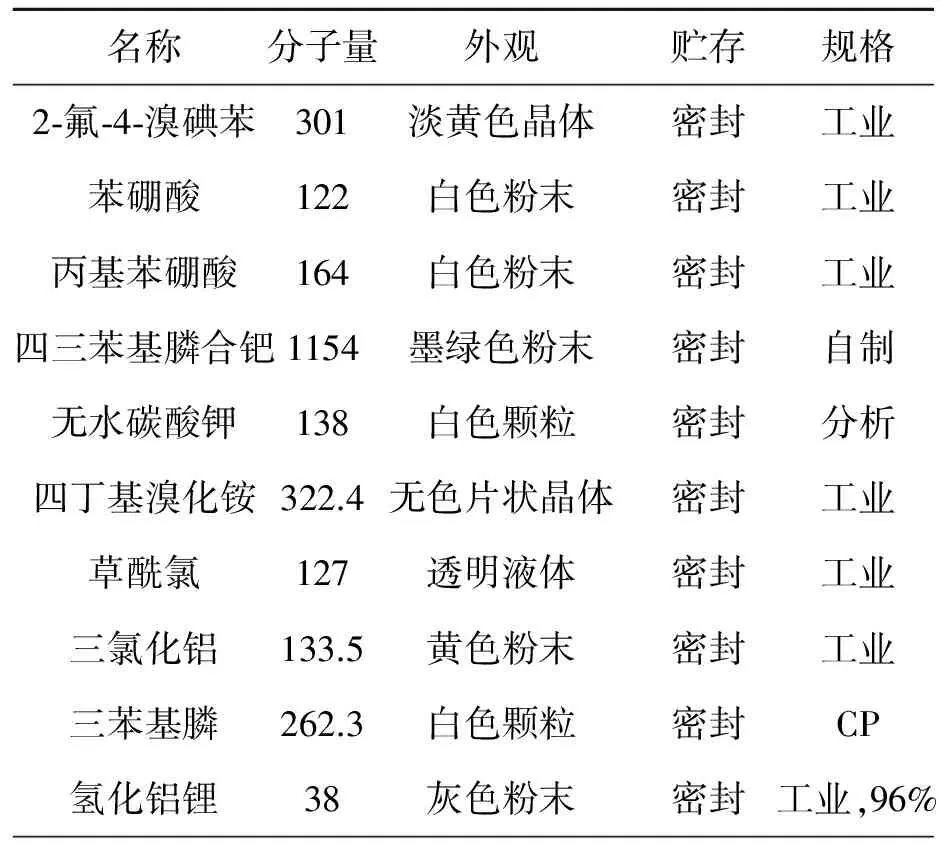
表1 原材料常数表

续表
2.2 仪器与设备
仪器与设备见表2。
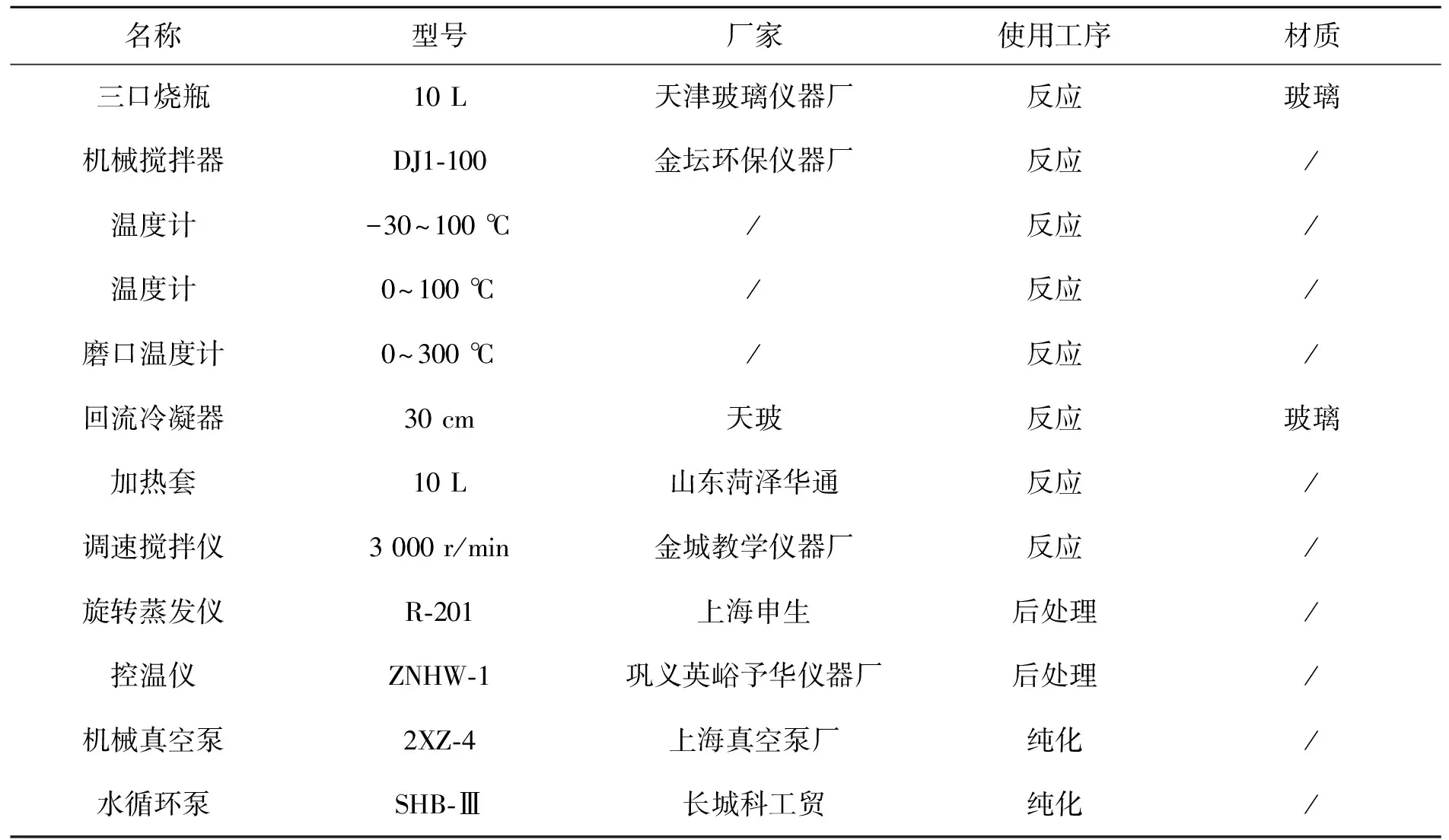
表2 仪器与设备
2.3 实验
2.3.1 化合物1 的合成
反应方程式:
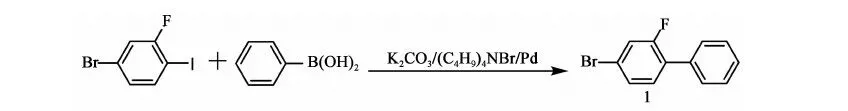
在氮气保护下,搅拌下向10 L三口烧瓶中加入2.1 kg甲苯、2.4 kg乙醇、800 g(2.66 mol)2-氟-4-溴碘苯、214.2 g(0.66 mL)四丁基溴化铵、356.7 g(2.9 mol)苯硼酸、12.27 g(0.011 mol)自制零价钯[Pd(Pph3)4]、0.4 kg水、777.6 g(5.63 mol)碳酸钾,加料毕,加热至回流(70~75 ℃)反应12 h,2-氟-4-溴碘苯GC<0.5%,停止反应。降至室温,将反应液倒入盛有7 kg水和1.9 kg甲苯的容器中,搅拌10 min,静置,分液,保留有机相,水相用1.74 kg甲苯提取一次,合并有机相,用8 kg×3的水水洗至中性。有机相用200 g无水硫酸镁干燥,过滤,滤饼用0.22 kg×2甲苯淋洗,滤液合并,浓缩(真空度>0.085 MPa,70~80 ℃),得褐色液体648.5 g,粗品收率97%。高真空蒸馏,收集110~112 ℃/50 Pa下馏分,得466 g油状物化合物1:2-氟-4-溴联苯,GC含量99.49%,收率70%(以2-氟-4-溴碘苯计)。
2.3.2 化合物2的合成
反应方程式:

在干燥的带有尾气吸收装置的10 L三口瓶中加入8.6 kg二氯甲烷,搅拌下加入860 g(6.47 mol)三氯化铝,降温至0~5 ℃,加入650 g(2.59 mol)化合物1,搅拌15 min,滴加658 g(5.18 mol)草酰氯,约30 min滴完,整个滴加过程温度控制在0~5 ℃。滴毕,升温至10~15 ℃反应1 h后取样分析,当化合物GC含量<0.5%时,停止反应,将反应液缓慢倒入装有2.66 kg二氯甲烷,0 ℃左右稀盐酸(VHCl:VH2O=1∶3)的容器中,搅拌15 min,静置,分液,有机相待用,水相用二氯甲烷提取一次,合并有机相,以8 kg×3的水,水洗至中性。有机相以300 g无水硫酸镁干燥,过滤,滤饼用少量二氯甲烷淋洗,合并有机相,浓缩(真空度>0.08 MPa,水温70~80 ℃),烘料(0.095 MPa,45 ℃,4 h),得黄色固体化合物2:2-氟-4-溴-1-乙酰氯基联苯 769 g,GC含量94.3%,mp.104~112 ℃),收率94.7%(以化合物1计)。
2.3.3 化合物3的合成
反应方程式:
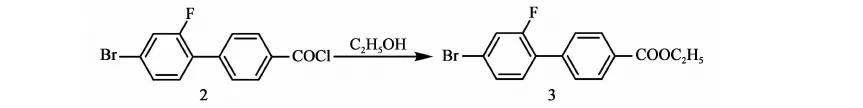
在干燥的,带有尾气吸收装置的10 L三口瓶中加入5.2 kg甲苯,1 200 g(3.83 mol)化合物2,搅拌10 min(体系橘红色浑浊),加入522 g(11.35 mol)乙醇,升温至回流(78~80 ℃)反应,当化合物2 GC含量<0.5%时,停止反应。降温至50 ℃以下,将反应液缓慢倒入装有0.87 kg甲苯和2.4 kg水的容器中,搅拌10 min,静置15 min,分液,有机相待用,水相用1.2 kg甲苯提取一次,搅拌10 min,静置,分液,合并有机相,水洗至中性。有机相用200 g无水硫酸镁干燥,过滤,滤饼用少量甲苯淋洗,合并有机相,浓缩(真空度>0.085 MPa,水温70~80 ℃),得红褐色液体化合物3:2-氟-4-溴-1-乙酸乙酯基联苯1 224 g,GC含量96.1%,单步收率99%(以化合物2计)。
2.3.4 化合物4的合成
反应方程式:
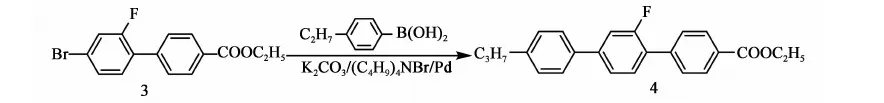
在氮气保护下,搅拌下向10 L三口烧瓶中依次加入2.2 kg甲苯、845 g(2.62 mol)化合物3,472 g(2.88 mol)丙基苯硼酸、2.57 kg乙醇、211 g(0.65 mol)四丁基溴化铵、12.1 g(0.01 mol)零价钯、422.5 g水、765.5 g(5.55 mol)碳酸钾,加料完毕,加热至回流(70~75 ℃)反应,当原料 GC含量<0.5%时,停止反应。降温至50 ℃以下,后处理。将反应液倒入盛有7 kg水和2.5 kg甲苯的容器中,搅拌10 min,静置,分液,保留有机相,水相用1.7 kg甲苯提取一次,合并有机相,用水洗至中性。有机相用200 g无水硫酸镁干燥,过滤,滤饼用少量甲苯淋洗,滤液合并浓缩(真空度>0.085 MPa,70~80 ℃),得灰褐色固体917.5 g,粗品收率96.9%(以化合物3计)。向上述产品加入0.8 kg甲苯,0.82 kg乙醇,加热至70~80 ℃,溶解后,降至室温后,冷冻(-20 ℃,12 h),过滤,重复上述直至主含量GC>99%,产品烘干(0.095 MPa,70~80 ℃,8 h),得产品化合物4:4-乙酸乙酯基-2′-氟-4″丙基-[1,1′,4′1″]三联苯:707.5 g,GC含量99.6%,mp.=160~163 ℃),收率74.7%(以化合物3计)。
2.3.5 化合物5的合成
反应方程式:
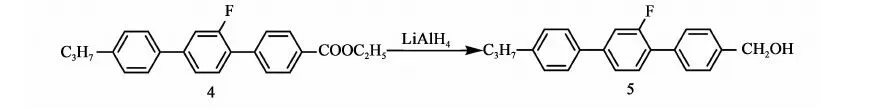
向10 L三口瓶中通氮气5 min,加入1.16 kg四氢呋喃,然后分批加入48.8 g(1.28 mol)氢化铝锂,加料完毕,搅拌10 min后开始滴加636 g(1.76 mol)化合物4和1.7 kg四氢呋喃的混合液,控温55~65 ℃,约30 min滴完,滴完升温至回流(60~65 ℃),保温30 min后取样分析,当原料GC含量<0.1%时,停止反应,后处理。体系降温至35~45 ℃,向体系中滴加0.08 kg丙酮,搅拌10 min向体系中加入1.74 kg甲苯,再滴加500 ml稀盐酸(250 mL浓盐酸+250 mL水),待体系温度降至30~40 ℃时,倒入20 L桶中。向体系中加入1.7 kg甲苯,7 kg水搅拌10 min,静置,分液,有机相待用,水相用1.7 kg甲苯提取,合并有机相,用水洗至中性,有机相用250 g无水硫酸镁干燥,过滤,滤饼用0.174 kg×2的甲苯淋洗,合并有机相浓缩(真空度>0.085 MPa,70~80 ℃),得黄色固体530 g,粗品收率94.3%。向产品中加入甲苯(1 g粗品∶3 mL甲苯),加热至75~80 ℃,溶解后,冷冻(-20 ℃,12 h),过滤,重复上述操作直至GC>99%,烘干(0.09 MPa,40~50 ℃,8 h),得产品化合物5:4-甲醇基-2′-氟-4″丙基-[1,1′,4′1″]三联苯:460 g,GC含量99.3%,mp.=121.5~123.4 ℃),收率81%(以化合物4计)。
2.3.6 化合物6的合成
反应方程式:
搅拌下,向干燥的1 L三口瓶中依次加入4.85 kg甲苯,930 g (2.9 mol)化合物5,277 g(3.5 mol)吡啶,加热,待体系温度50~55 ℃,开始滴加415 g(3.5 mol)氯化亚砜与0.36 kg甲苯的混合溶液,控温60~70 ℃,约60 min滴完,70~75 ℃下保温反应1 h,取样分析,当原料LC含量<0.1%时,停止反应。降温至50 ℃以下,静置2 h后,分液,上层有机相在搅拌下倒入盛有1.25 kg甲苯,50 g氯化钠,3 L稀盐酸(1 500 mL浓盐酸+1 500 mL水)的容器中,搅拌10 min,静置,分液,有机相待用,水相用甲苯提取一次,合并有机相,水洗至中性,有机相用300 g无水硫酸镁干燥,过滤,滤饼用甲苯淋洗,合并滤液浓缩(真空度>0.085 MPa,70~80 ℃),得粗品877 g,粗品收率89.1%(以化合物5计)。将上述粗品以1 g粗品∶2 mL甲苯:1 mL乙醇重结晶(-20 ℃,8 h),至LC>99.5%,得产品化合物6:4-氯甲基-2′-氟-4″丙基-[1,1′,4′1″]三联苯:650 g,mp.=112.9~114.7),收率66.7%(以化合物5计)。
2.3.7 目标产物TM的合成
(1)氯丙烯格氏试剂的制备
反应方程式:
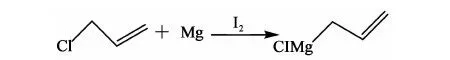
在氮气保护下,搅拌下向10 L三口瓶中依次加入31 g四氢呋喃、33.4 g(1.4 mol)镁粉,滴入少量氯丙烯使其引发(体系温度60~70 ℃),滴加67.5 g(0.88 mol)氯丙烯,滴加过程中控制体系温度在-10~0 ℃,约35 min滴完。滴毕在-10~0 ℃下反应0.5 h,室温静置1 h,将上层清液倒入1 L三口瓶中,密封待用。
(2)化合物TM的制备
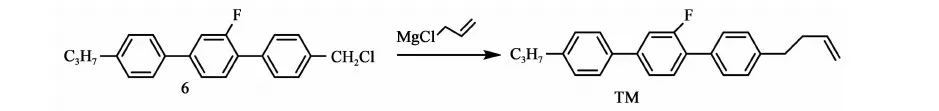
氮气保护下,将上述格氏试剂升温至40 ℃,滴加60 g(0.18 mol)化合物6与0.24 kg四氢呋喃的混合溶液,滴加过程中温控40~50 ℃,滴完保温反应1 h,当原料LC含量<0.05%时,停止反应。将反应液倒入盛有230 ml稀盐酸(80 mL浓盐酸+150 mL水),0.26 kg甲苯的烧杯中,搅拌15 min,静置,分液,有机相待用,水相用60 mL甲苯提取一次,合并有机相,用水洗至中性,有机相用20 g无水硫酸镁干燥,过滤,滤饼用60 mL甲苯淋洗一次,合并滤液浓缩(真空度>0.085 MPa,70~80 ℃),得粗品63 g,粗品收率103%。将上述粗品以1 g粗品∶2 mL乙醇重结晶,过滤,滤饼用15 g乙醇淋洗,烘干(0.095 MPa,35~40 ℃,4 h),得目标产物4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′,1″]三联苯的合成:50 g,LC含量99.9%;GC含量99.8%,收率81.9%(以化合物6计)。IR(v/cm-1):3 023,2 955,2 925,2 866,1 915,1 639(C=C),1 546,1 487,1 396,1 182,1 132,1 005,893,798;1H NMR(CDCl3,500 MHz):7.41~7.53(m,7H),7.23~7.38(m,4H),5.06~5.09(d,J=18.5 Hz,1H),4.99~5.01(d,J=10.5 Hz,1H),2.74~2.77 (t,J=7.75 Hz,2H),2.61~2.64 (t,J=7.75 Hz,2H),2.39~2.44(m,2H),1.64~1.72(m,2H),0.96~0.98(t,J=7.25 Hz,3H); MS(m/z %):344.5(M+,40),303(100),274(40)。
总收率:21.7%(以2-氟-4-溴碘苯计)。
3 结果与讨论
3.1 化合物1的合成讨论
本步反应重复3次,3次反应中控取样分析结果见表3。
表3化合物1反应时间与产物含量变化表
Tab.3 Relation between content of compound 1 and reaction time

反应名称18h产物GC含量%原料GC含量%20h产物GC含量%原料GC含量%22h产物GC含量%原料GC含量%反应一//93.91.793.40.2反应二91.71.594.60.09//反应三94.41.3091.90.04//
以上数据看出,该反应在20~22 h可终止反应。
3.2 化合物2的合成讨论
本步反应重复3次,3次反应取样分析结果见表4。
表4化合物2反应时间与产物含量变化表
Tab.4 Relation between content of compound 2 and reaction time
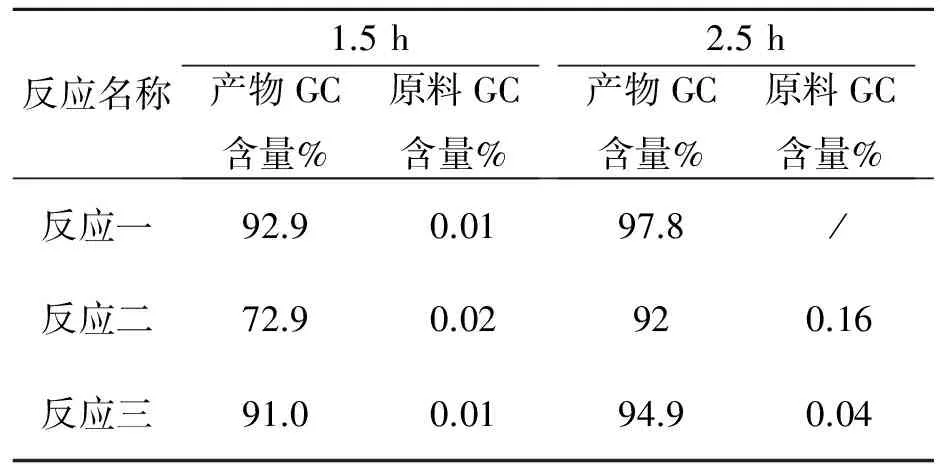
反应名称1.5h产物GC含量%原料GC含量%2.5h产物GC含量%原料GC含量%反应一92.90.0197.8/反应二72.90.02920.16反应三91.00.0194.90.04
以上数据看出本反应在2.5 h可停止反应。
3.3 化合物3的合成讨论
本步反应重复3次,3次反应中取样分析结果见表5。
表5化合物3反应时间与产物含量变化表
Tab.5 Relation between content of compound 3 and reaction time
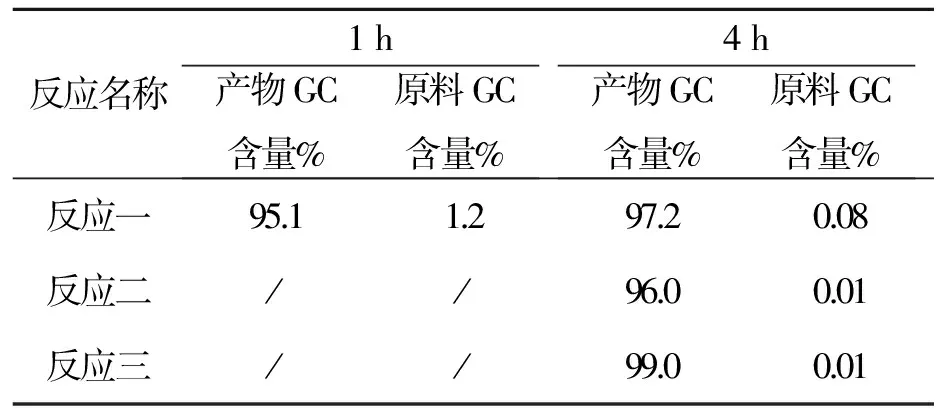
反应名称1h产物GC含量%原料GC含量%4h产物GC含量%原料GC含量%反应一95.11.297.20.08反应二//96.00.01反应三//99.00.01
以上数据看出本反应重复性好,在4 h停止结束。
3.4 化合物4的合成讨论
本步分别用钯炭(反应一,二)和零价钯(反应三,四)2种体系,取样分析结果见表6。
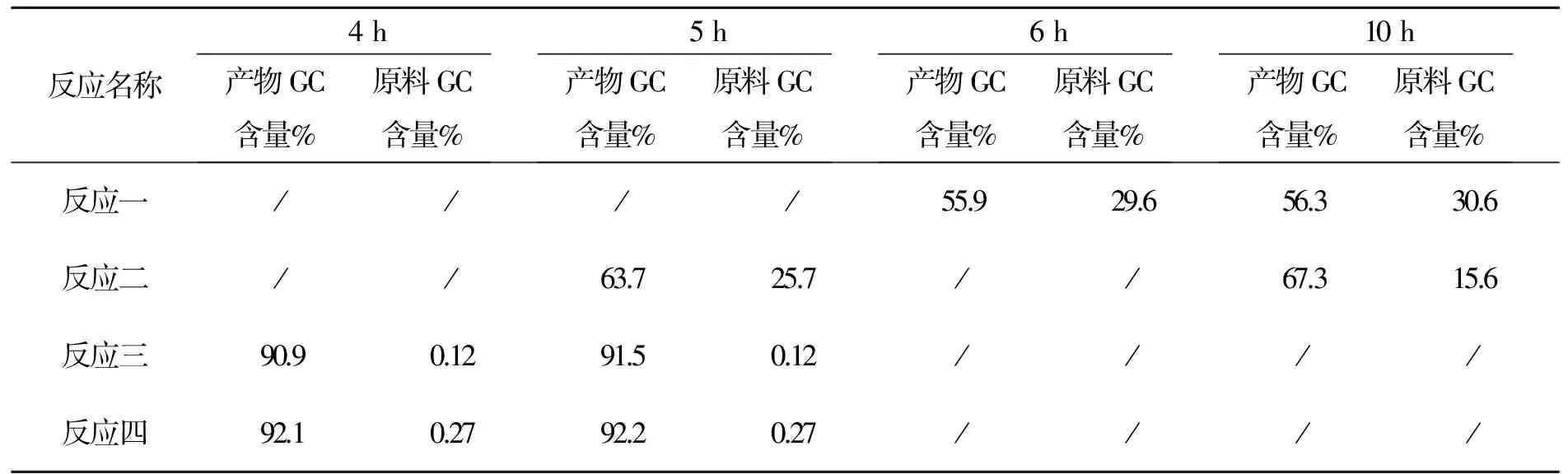
表6 化合物4反应时间与产物含量变化表
反应一:钯炭体系∶硼酸=1∶1.1;反应二钯炭体系∶硼酸=1∶1.8;反应三:四零价钯体系∶硼酸=1∶1.1。
以上数据可以看出本反应零价钯体系4 h可以反应完全,钯炭体系硼酸比例为1∶1.8仍反应不完。
3.5 化合物5的合成讨论
本步反应取样分析结果见表7。
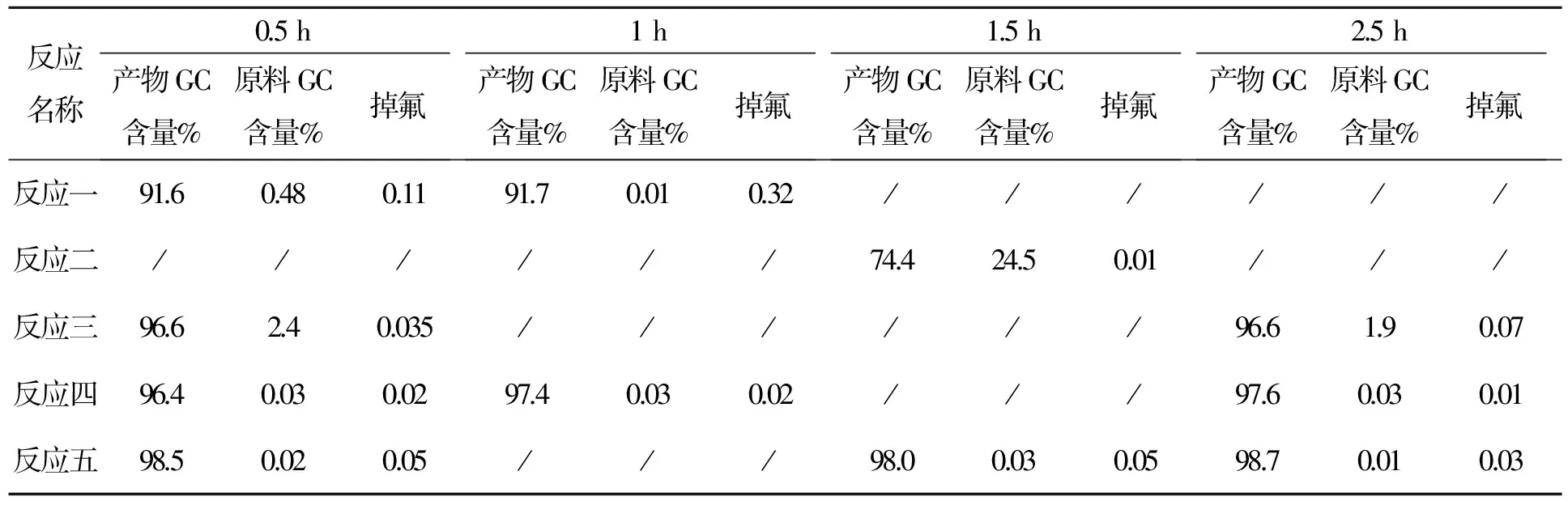
表7 化合物5反应时间与产物含量变化表
各组氢化铝锂料比,反应一1∶1.2;反应二1∶0.5;反应三1∶0.6;反应四五1∶0.7。从以上数据可以看出氢化铝锂比例1∶1.2时,产生掉氟较大,当氢化铝锂比例为1∶0.5、1∶0.6时,原料有剩余;当氢化铝锂比例为1∶0.7时1 h可反应完全,掉氟<0.05%。
3.6 化合物6的合成讨论
本步反应取样分析结果如下
各组实验料比:化合物5∶SOCl2∶吡啶
反应一 1∶1∶0
反应二 1∶1∶0.3
反应三 1∶1∶1
反应四 1∶1.2∶1(4 h后补加20%氯化亚砜)
反应五 1∶3∶1.2
反应六 1∶1.2∶1.2(4 h后补加20%氯化亚砜,5 h后二次补加20%氯化亚砜)
分析结果见表8。
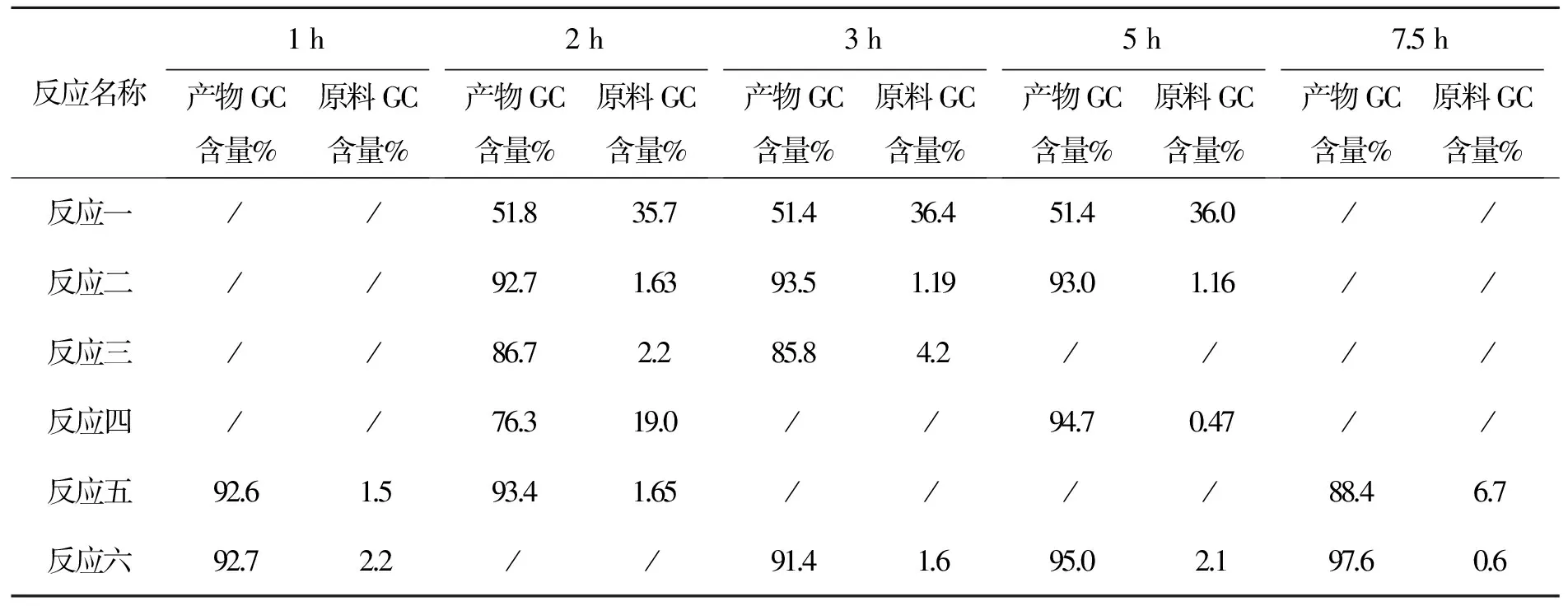
表8 化合物6反应时间与产物GC含量变化表
以上数据看出:(1)吡啶加快了反应速度。(2)当氯化亚砜调至1∶3时延长反应时间仍反应不完。(3)氯化亚砜需多次补加才能使原料反应完全,采用LC进行跟踪,数据见表9。
表9化合物6反应时间与产物LC含量变化表
Tab.9 Relation between content of compound 6 and reaction time
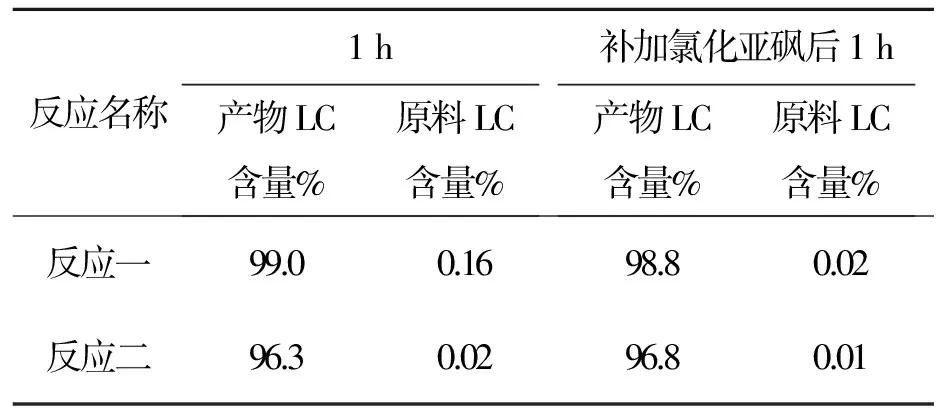
反应名称1h产物LC含量%原料LC含量%补加氯化亚砜后1h产物LC含量%原料LC含量%反应一99.00.1698.80.02反应二96.30.0296.80.01
以上数据看出,用LC跟踪补加氯化亚砜后原料可反应完全。
3.7 目标产物TM的合成讨论
本步温度条件反应重复两次,两次反应中取样分析结果见表10。
表10化合物7反应时间与产物GC含量变化表
Tab.10 Relation between content of compound 7 and reaction time
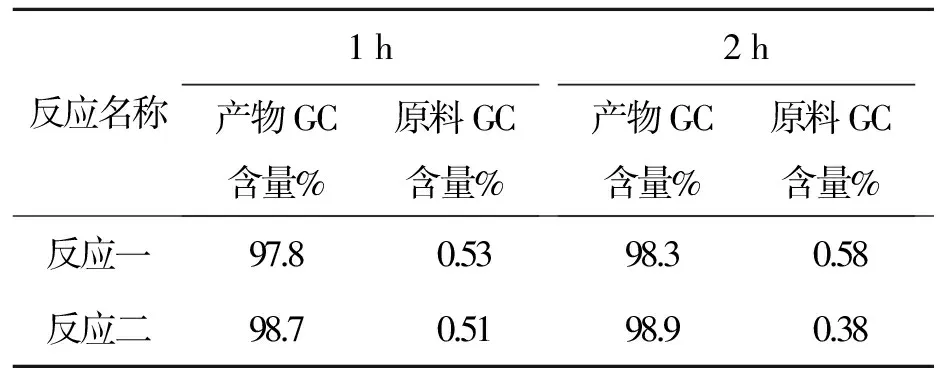
反应名称1h产物GC含量%原料GC含量%2h产物GC含量%原料GC含量%反应一97.80.5398.30.58反应二98.70.5198.90.38
以上数据可以看出控温35~40 ℃,原料反应至0.5%左右很难再减小。故把温度升高,改为40~45 ℃,且用GC//LC对比,数据见表11。
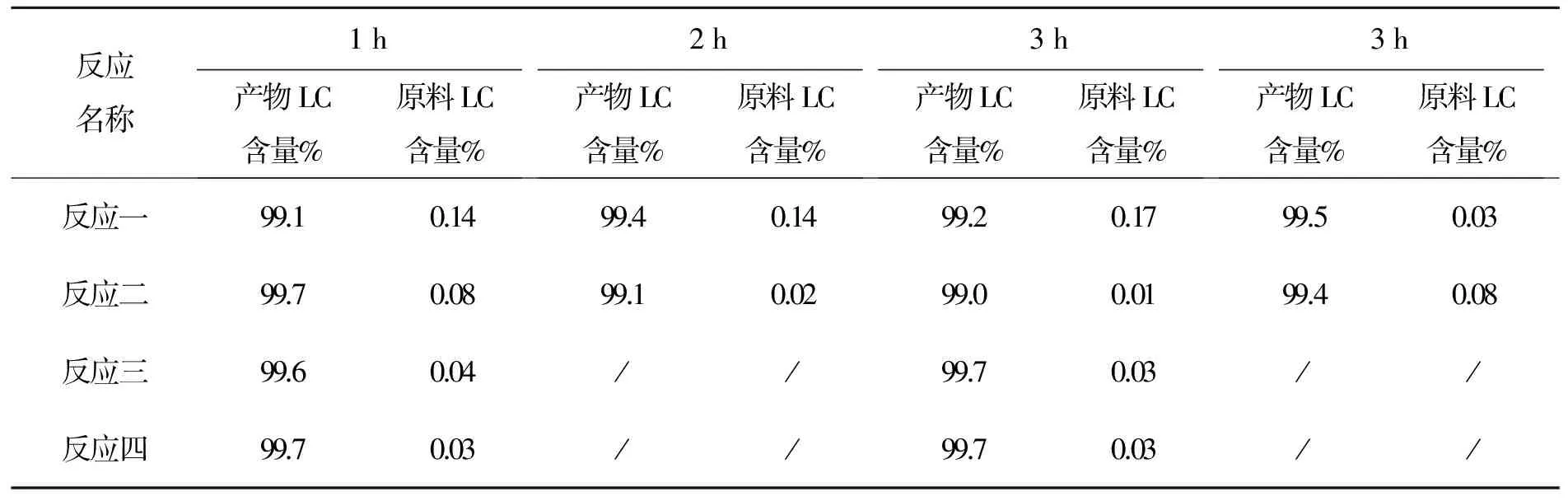
表11 化合物7反应时间与产物LC含量变化表
从以上数据看出,该反应用LC跟踪更精确,3 h可停止反应。
4 结 论
本文以2-氟-4-溴碘苯和苯硼酸为原料,共经过7步反应得到目标产物,总收率21.7%。与底物不同的类似文献路线相比,首次路线完整的报道了4-(3-烯)正丁基-2′氟-4″丙基-[1,1′,4′,1″]三联苯的合成过程。本文路线与文献报道的构架丁烯端基方法一相比较,相似之处在于都进行了苄醇结构、苄氯结构的合成以及与烯丙基氯格氏试剂的反应,不同之处在于苄醇结构的来源。文献通过锂试剂超低温反应水解得到苯甲醛基,然后还原制得苄醇,成本较高,且醛基易被氧化,不易保存;本文通过酰化、酯化得到苯甲酸酯基,然后还原制得苄醇,酯性能稳定,易于保存,且成本降低。与文献报道的构架丁烯端基方法二相比较,文献采用了苄溴基与烯丙基溴格氏试剂反应,同样采用锂试剂催化、超低温反应条件(-50 ℃),本文采用烯丙基氯格氏试剂反应,温度40 ℃左右,条件温和,易于操作。文中详细讨论了各反应过程中反应时间对产物转化的影响。与课题组开发的此化合物其它合成路线相比,最后一步构架丁烯端基,可较好的控制烯键易位,易纯化。反应过程所用原料廉价易得,可有效降低成本,同时利于实现工业化生产。
[1] 员国良,郑成武,华瑞茂. 含链端烯基负性液晶单体的合成及其性能研究[J].液晶与显示,2013,28(4):510-515,551.
Yun G L,Zheng C W,Hua R M.Preparation and characteristics of terminal alkeny bearing lateral fluoro benzene negative liquid crystal [J].ChineseJournalofLiquidCrystalsandDisplays,2013,28(4):510-515,551. (in Chinese)
[2] 赵地顺,李洪胜,段二红,等. 双烯含氟类液晶化合物的合成与表征[J].河北师范大学学报:自然科学版,2010,34(4):448-452.
Zhao D S,Li H S,Duan E H,etal.Synthesisand properties of diolefin fluorous liquid crystal compound [J].JournalofHebeiNormalUniversity:NaturalScienceEdition,2010,34(4):448-452.(in Chinese)
[3] 张婷婷,姜雪松,李永刚,等. 一种双乙烯类液晶化合物及其制备方法:中国,CN101928199 B[P]. 2010-08 -04.
Zhang T T,Jiang X S,Li Y G,etal.Divinyl liquid crystal compound and preparation method:China,CN101928199 B[P]. 2010-08-04.(in Chinese)
[4] 胡葆华,袁鹄,周银波,等.丁烯类化合物:中国,CN102203214 B [P].2013-07-24.
Hu B H,Yu H,Zhou Y B,etal. Butenes:China,CN102203214 B [P].2013-07-24.(in Chinese)
[5] 任惜寒,孟劲松,员国良,等.烷基双环己基2、3-二氟苯丁烯类液晶化合物及其用途:中国,CN102153442 A [P].2011-08-17.
Ren X H,Men J S,Yun G L,etal.Alkyl dicyclohexyl 2,3-difluoro-butene liquid crystal compounds and their use:China,CN102153442 A [P].2011-08-17.(in Chinese)
[6] KELLY S M. Four unit linking groups 3 liquid crystals of negetive dielectric anisotropy [J].LiquidCrystals,1991,10(2):261-272.
[7] KELLY S M.Four unit linking groups 4 Liquid crystals of negetive dielectric anisotropy [J].LiquidCrystals,1991,10(2):273-287.
[8] SHIMADA. Liquid crystalline compound,Liquid crystalcomposition,Liquid crystaldisplay element,WO2008090780 A1[P].2008-07-31.
[9] 员国良,贾刚刚,梁志安,等. 含有双3-丁烯基含氟三联苯液晶化合物及其制备方法:中国,CN201110051298[P]. 2011-03-04.
Yun G L,Jia G G,Liang Z A,etal.Double 3-butene containing fluorinated terphenyl liquid crystal compound and preparation method thereof:China,CN201110051298 [P]. 2011-03-04.(in Chinese)
[10] Rajiv R S,Robert R,Singhaus G W. 4-dihydroxyborylphenyl analogues of 1-aminocyclobutanecarboxylic acids:potential boron neutron capture therapy agents [J].J.Org.Chem,1999,64(23):8495-8500.
猜你喜欢
中老年保健(2022年3期)2022-08-24
数学物理学报(2021年6期)2021-12-21
中华养生保健(2020年9期)2021-01-18
化工设计通讯(2021年4期)2021-01-07
液晶与显示(2020年8期)2020-08-08
中国材料进展(2019年10期)2019-12-07
无机化学学报(2019年2期)2019-02-27
应用能源技术(2019年1期)2019-01-30
液晶与显示(2015年1期)2015-02-28
郑州大学学报(工学版)(2014年6期)2014-03-01
