
塞来昔布对戊四氮点燃腺苷A1受体敲除小鼠癫痫发作及PGP、COX-2表达的影响*
2014-01-01 09:26康慧聪张继龙刘志广朱遂强
华中科技大学学报(医学版) 2014年5期
单 萍, 康慧聪, 张继龙, 刘志广, 李 巷, 朱遂强△
1武汉市第一医院神经内科,武汉 430022
2华中科技大学同济医学院附属同济医院神经内科,武汉 430030
研究发现,癫痫发作可导致环氧合酶-2(cyclooxygenase-2,COX-2)表达上调[1];而癫痫动物脑内COX-2的激活又可促进癫痫活动诱导的神经元死亡,引起异常的神经轴、树突发芽和胶质增生等病理改变(难治性癫痫脑组织病理特点之一),缩短癫痫发作的潜伏期,从而促进癫痫的反复发作(难治性癫痫的临床表现特点之一)[2],这提示COX-2可能与癫痫耐药性产生有关。国外已有学者证实了COX-2 与 多 药 转 运 体 (multidrug transporters,MDTs)介导的难治性癫痫的相关性[3]。此外,COX-2通 过 COX-2/前 列 腺 素 E2(prostaglandin E2)通路产生大量PGE2,激活相关受体,极大促进胶质细胞增殖及神经元可塑性改变[4-5],而胶质细胞活化增殖又可导致腺苷系统功能障碍从而促进COX-2/PGE2通路的反应[6],形成恶性循环。因此我们推测COX-2诱导癫痫耐药性产生的机制中极有可能有腺苷能系统的参与。本研究通过观察塞来昔布(选择性COX-2抑制剂)对戊四氮(pentylenetetrazol,PTZ)点燃腺苷A1受体敲除小鼠P-糖蛋白 (P-glycoprotein,PGP)、COX-2 的 表 达 来 探 讨COX-2与癫痫耐药性产生的关系及其可能机制。
1 材料与方法
1.1 实验动物
腺苷A1受体基因敲除小鼠50只,6~8周龄,体重20~25g。种鼠由美国国立糖尿病消化病与肾病研究所Schnermann教授赠送,配种后自然繁殖,饲养于华中科技大学同济医学院附属同济医院实验动物中心SPF级屏障系统,室温18~22℃,每日12 h光照,普通饮食,IVC-Ⅱ型(智能型)独立送风隔离笼具。
1.2 分组
小鼠随机分为3组。对照组:10只,不给予PTZ点燃;非干预组:20只,PTZ点燃前30min腹腔注射等量生理盐水,再分为非干预24h亚组及非干预30d亚组,各10只;干预组:20只,PTZ点燃前30min腹腔注射塞来昔布溶液(10mg/kg),再分为干预24h亚组及干预30d亚组,各10只。
1.3 主要仪器设备和试剂
基因扩增仪(PTC-1160),Bio-Rad公司;电泳仪(DYY-6B),北京市六一仪器厂;凝胶成像系统,Gene Bio Imaging System;台式冷冻离心机,Gene Company Limited;冰冻切片机(CM1900),德国莱卡公司;荧光显微镜 OLYMPUS(DP50-CU),日本OLYMPUS公司;电热恒温水浴锅(DK-S24型),上海精宏实验设备有限公司。
蛋白酶 K,德国 Merck公司;2×Taq PCR MasterMix、TRNzol,天根生化科技(北京)有限公司;琼脂糖,西班牙;无水乙醇、氯化钠、盐酸(均为分析纯),上海振兴化工厂;Bio MarkerⅠ,BioFlux公司;引物设计及合成,上海Invitrogen生物工程公司;goldview,赛百威公司;PTZ,美国Sigma公司;塞来昔布(celecoxib,西乐葆),辉瑞制药有限公司(生产批号:BH20050352);逆转录 RT试剂盒,TOYOBO(日本);多药耐药(multi-drug resistance,MDR)1基因兔抗羊多克隆抗体、COX-2兔抗羊多克隆抗体、荧光 FITC标记试剂(FITC-Avidin)、SABC-Cy3免疫组化试剂盒(生物素化二抗:抗兔IgG、SABC-Cy3),武汉博士德生物工程有限公司;冷冻切片包埋剂(OTC),中杉金桥公司。
1.4 PTZ点燃模型的制备及行为学观察
对照组给予等量生理盐水腹腔注射,干预组和非干预组均每日腹腔注射PTZ(30mg/kg)直到点燃成功;若连续每日腹腔注射PTZ 14d仍未点燃者,不再继续注射,为点燃不成功。每次注射PTZ后1h内观察各组小鼠行为,记录癫痫发作情况。癫痫惊厥发作评分标准(按Smialowki癫痫发作分级标准):0级,无反应;1级,面部运动,指鼻子、点头、奔跑;2级,前肢痉挛;3级,前肢痉挛伴后退;4级,前肢痉挛伴后退及摔倒发作;5级,摔倒、强直发作。点燃成功标准:连续3dPTZ腹腔注射后,均表现为4~5级发作,为点燃成功。观察并记录各组小鼠PTZ点燃过程中点燃潜伏时间、发作开始时间、发作持续时间。
1.5 RT-PCR法检测各组小鼠大脑皮层和海马PGP、COX-2 mRNA的表达
采用脊椎脱臼法处死小鼠,称取50mg皮层(或海马)脑组织,用TRNzol试剂提取RNA。取1 μL RNA溶解于99μL的无RNA酶水中,紫外线分光光度仪上分别读取在260nm和280nm波长处的吸光度(A)值,测定RNA浓度,得到A260/A280比值,比值位于1.8~2.0之间。采用TOYOBO公司的逆转录试剂盒合成cDNA。PGP cDNA(mdr1)基因全长从Pubmed GenBank中查得,由上海Invitrogen生物工程公司设计并合成,β-actin序列从有关数据中获得并在GenBank中进行核对,各自序列见表1。mdr1PCR反应体系:2×Taq PCR MasterMix 8μL,ddH2O 6μL,上下游引物各0.5μL,1 μL cDNA,总反应体积16μL;PCR反应条件:mdr1循环参数为94℃预变性8min,94℃变性30s,57℃退火30s,72℃延伸45s,36个循环后,72℃后延伸8min,4℃冰箱保存。COX-2PCR反应体系:2×Taq PCR MasterMix 10μL,ddH2O 7μL,上下游引物各 0.5μL,cDNA 2μL,总反应体积 20μL。COX-2循环参数为:94℃预变性5min,94℃变性21s,55℃退火21s,72℃延伸30s,30个循环后,72℃后延伸8min,4℃冰箱保存。PCR反应产物在1.5%琼脂糖凝胶上电泳,使用美国Gene Bio凝胶电泳成像系统测出目的基因和β-actin的表达强度,以同一标本的β-actin的产物积分A值校正各自目的基因的积分A值(目的基因表达相对值=目的基因A 值/β-actin A 值)。

表1 PGP cDNA、COX-2cDNA基因序列Table 1 Gene sequences of PGP and COX-2cDNA
1.6 免疫荧光组织化学染色法检测小鼠大脑皮层和海马PGP、COX-2蛋白的表达
取出切好后的冰冻切片,放入4%多聚甲醛中浸泡15min(4℃,固定);弃多聚甲醛,加入磷酸盐缓冲液(PBS,pH 值7.2~7.4)摇床洗涤3次(摇床60次/min),每次5min;洗后甩干,加封闭液2h;甩干,加入mdr1兔抗羊多克隆抗体或COX-2兔抗羊多克隆抗体(一抗)(1∶100),4℃孵育过夜;PBS摇床洗涤3次,每次5min,甩干,加入抗兔IgG(二抗)(1∶100),室温孵育1h;PBS摇床洗涤3次,每次5 min,甩干,加入Cy3或FITC-Avidin(1∶100),室温孵育1h;自来水冲洗45min,甩干,待组织完全干后,加入50%甘油封片。阴性对照:以PBS代替一抗,同时进行上述免疫荧光组织化学染色,结果为阴性。在荧光显微镜下(×400),对各组小鼠脑片免疫荧光组织化学染色阳性细胞进行计数。每1张脑片在皮层(或海马)随机选取6个视野,求其平均值。
1.7 统计学分析
应用SPSS 15.0统计软件处理数据,计量资料采用均数±标椎差(±s)表示,组间均数比较采用方差分析,以P<0.05为差异有统计学意义。
2 结果
2.1 行为学观察
各组小鼠PTZ点燃过程中点燃潜伏时间、发作开始时间、发作持续时间比较结果显示:PTZ点燃过程中,对照组未出现癫痫发作;干预组点燃潜伏时间及发作开始时间均长于非干预组(P<0.05),发作持续时间短于非干预组(P<0.05),见表2。
表2 各组小鼠点燃潜伏时间、发作开始时间、发作持续时间比较(±s)Table 2 Comparison of the kindling latent time,start time of seizures,and duration of seizures in the mice among groups(±s)

表2 各组小鼠点燃潜伏时间、发作开始时间、发作持续时间比较(±s)Table 2 Comparison of the kindling latent time,start time of seizures,and duration of seizures in the mice among groups(±s)
与非干预组比较,*P<0.05
组别 n 点燃潜伏时间(d) 发作开始时间(min) 发作持续时间(min)对照组10 0 0 0非干预组 20 2.56±1.49 6.28±1.35 20.15±6.23干预组 20 5.38±1.27* 10.72±2.03* 15.01±4.25*
2.2 PGP mRNA在各组小鼠脑内皮层和海马的表达
对照组皮层和海马内可见少量PGP mRNA的表达;点燃后24h,非干预24h亚组小鼠大脑皮层和海马PGP mRNA表达高于对照组(P<0.05),而干预24h亚组与对照组比较差异无统计学意义;点燃后30d时,两实验组小鼠大脑皮层和海马PGP mRNA的表达均高于对照组及24h亚组(P<0.05);PTZ点燃后同一时间点(点燃后24h和点燃后30d)干预组PGP mRNA的表达显著低于非干预组(P<0.05)。见表3、图1。
表3 各组小鼠皮层及海马PGP mRNA的表达比较(±s,n=5)Table 3 Comparison of the expression of PGP mRNA in the cortex and hippocampus among groups(±s,n=5)
与对照组比较,*P<0.05;与同组24h亚组比较,▲P<0.05;与同时间点非干预组比较,△P<0.05
脑区 对照组亚组皮层 0.94±0.04 1.39±0.06* 3.28±0.07*▲ 0.98±0.05△ 1.85±0.08非干预组非干预24h亚组 非干预30d亚组干预组干预24h亚组 干预30d*▲△海马 0.88±0.05 0.98±0.03* 2.17±0.08*▲ 0.84±0.04△ 1.19±0.06*▲△
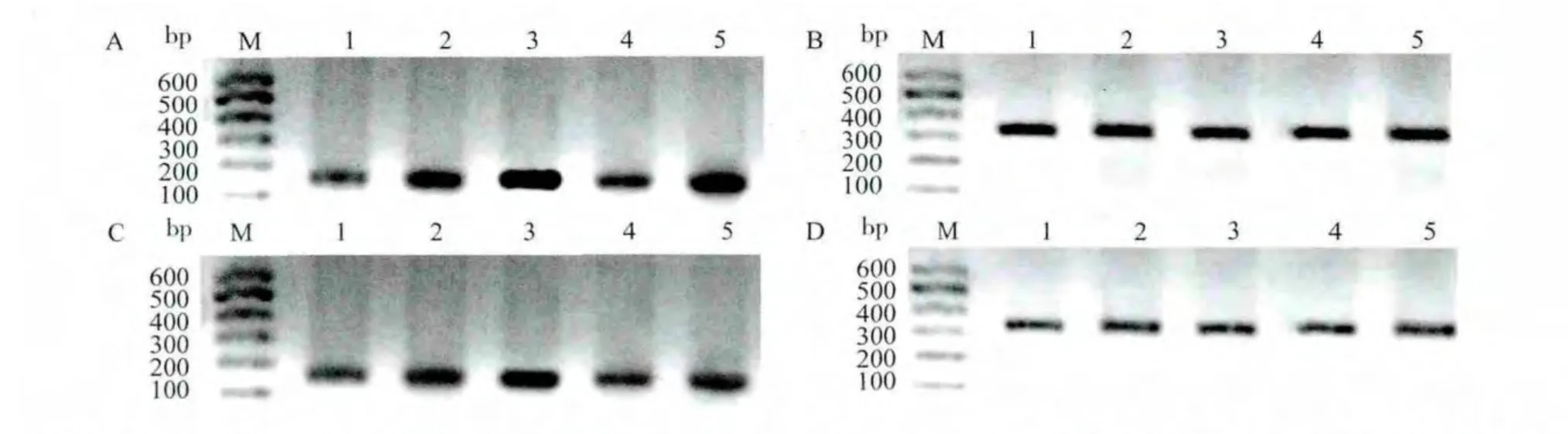
图1 PGP mRNA在各组小鼠脑内皮层和海马的表达Fig.1 Expression of PGP mRNA in the cortex and hippocampus of mice in each group
2.3 COX-2mRNA在各组小鼠脑内皮层和海马的表达
对照组皮层和海马内可见少量COX-2mRNA的表达;点燃后24h时,非干预24h亚组小鼠大脑皮层和海马COX-2mRNA表达高于对照组(P<0.05),而干预24h亚组与对照组比较差异无统计学意义;点燃后30d时,两实验组小鼠大脑皮层和海马COX-2mRNA的表达高于对照组及24h亚组(均P<0.05);PTZ点燃后同一时间点(点燃后24 h、点燃后30d)干预组COX-2mRNA的表达显著低于非干预组(均P<0.05)。见表4、图2。
2.4 PGP蛋白在各组小鼠脑内皮层和海马的表达
对照组皮层和海马内有少量细胞表达PGP,以胶质细胞阳性表达为主。点燃后24h时,非干预24 h亚组小鼠大脑皮层和海马PGP阳性细胞数高于对照组(P<0.05),而干预24h亚组与对照组比较差异无统计学意义;点燃后30d时,两实验组小鼠大脑皮层和海马PGP阳性细胞数高于对照组及24 h亚组(均P<0.05);PTZ点燃后同一时间点(点燃后24h、点燃后30d)干预组PGP阳性细胞数低于非干预组(均P<0.05)。见表5、图3。
表4 各组小鼠皮层及海马COX-2mRNA的表达比较(±s,n=5)Table 4 Comparison of the expression of COX-2mRNA in the cortex and hippocampus of mice among groups(±s,n=5)

表4 各组小鼠皮层及海马COX-2mRNA的表达比较(±s,n=5)Table 4 Comparison of the expression of COX-2mRNA in the cortex and hippocampus of mice among groups(±s,n=5)
与对照组比较,*P<0.05;与同组24h亚组比较,▲P<0.05;与同时间点非干预组比较,△P<0.05
脑区 对照组亚组皮层 0.77±0.05 0.96±0.04* 2.36±0.11*▲ 0.72±0.07△ 1.54±0.12非干预组非干预24h亚组 非干预30d亚组干预组干预24h亚组 干预30d*▲△海马 0.63±0.03 1.24±0.08* 2.87±0.09*▲ 0.67±0.06△ 1.56±0.10*▲△

图2 COX-2mRNA在各组小鼠脑内皮层和海马的表达Fig.2 Expression of COX-2mRNA in the cortex and hippocampus of mice in each group
表5 各组小鼠皮层及海马PGP阳性细胞数比较(±s,n=5)Table 5 Comparison of the number of PGP-positive cells in the cortex and hippocampus among groups(±s,n=5)

表5 各组小鼠皮层及海马PGP阳性细胞数比较(±s,n=5)Table 5 Comparison of the number of PGP-positive cells in the cortex and hippocampus among groups(±s,n=5)
与对照组比较,*P<0.05;与同组24h亚组比较,▲P<0.05;与同时间点非干预组比较,△P<0.05
脑区 对照组亚组皮层 9.17±1.03 15.82±1.97* 36.54±2.84*▲ 10.01±0.94△ 21.13±2.14非干预组非干预24h亚组 非干预30d亚组干预组干预24h亚组 干预30d*▲△海马 8.14±1.18 17.38±1.45* 39.18±2.16*▲ 8.81±2.01△ 24.32±2.62*▲△

图3 各组小鼠脑内皮层和海马PGP蛋白阳性细胞检测结果(免疫荧光,×400)Fig.3 PGP-positive cells in the cortex and hippocampus of mice in each group(immunofluorescence,×400)
2.5 COX-2蛋白在各组小鼠脑内皮层和海马的表达
对照组皮层和海马内有少量细胞表达COX-2;点燃后24h时,非干预24h亚组小鼠大脑皮层和海马COX-2阳性细胞数高于对照组(P<0.05),而干预24h亚组与对照组比较差异无统计学意义;点燃后30d时,两实验组小鼠大脑皮层和海马COX-2阳性细胞数显著高于对照组及24h亚组(均P<0.05);PTZ点燃后同一时间点(点燃后24h、点燃后30d)干预组COX-2阳性细胞数低于非干预组(均P<0.05)。见表6、图4。
表6 各组小鼠皮层及海马COX-2阳性细胞数比较(±s,n=5)Table 6 Comparison of the number of COX-2-positive cells in the cortex and hippocampus among groups(±s,n=5)

表6 各组小鼠皮层及海马COX-2阳性细胞数比较(±s,n=5)Table 6 Comparison of the number of COX-2-positive cells in the cortex and hippocampus among groups(±s,n=5)
与对照组比较,*P<0.05;与同组24h亚组比较,▲P<0.05;与同时间点非干预组比较,△P<0.05
脑区 对照组亚组皮层 17.58±3.67 29.41±4.31* 50.03±3.86*▲ 18.24±3.14△ 34.09±4.12非干预组非干预24h亚组 非干预30d亚组干预组干预24h亚组 干预30d*▲△海马 15.43±4.17 35.41±6.32* 52.49±5.18*▲ 16.59±5.63△ 38.29±7.15*▲△
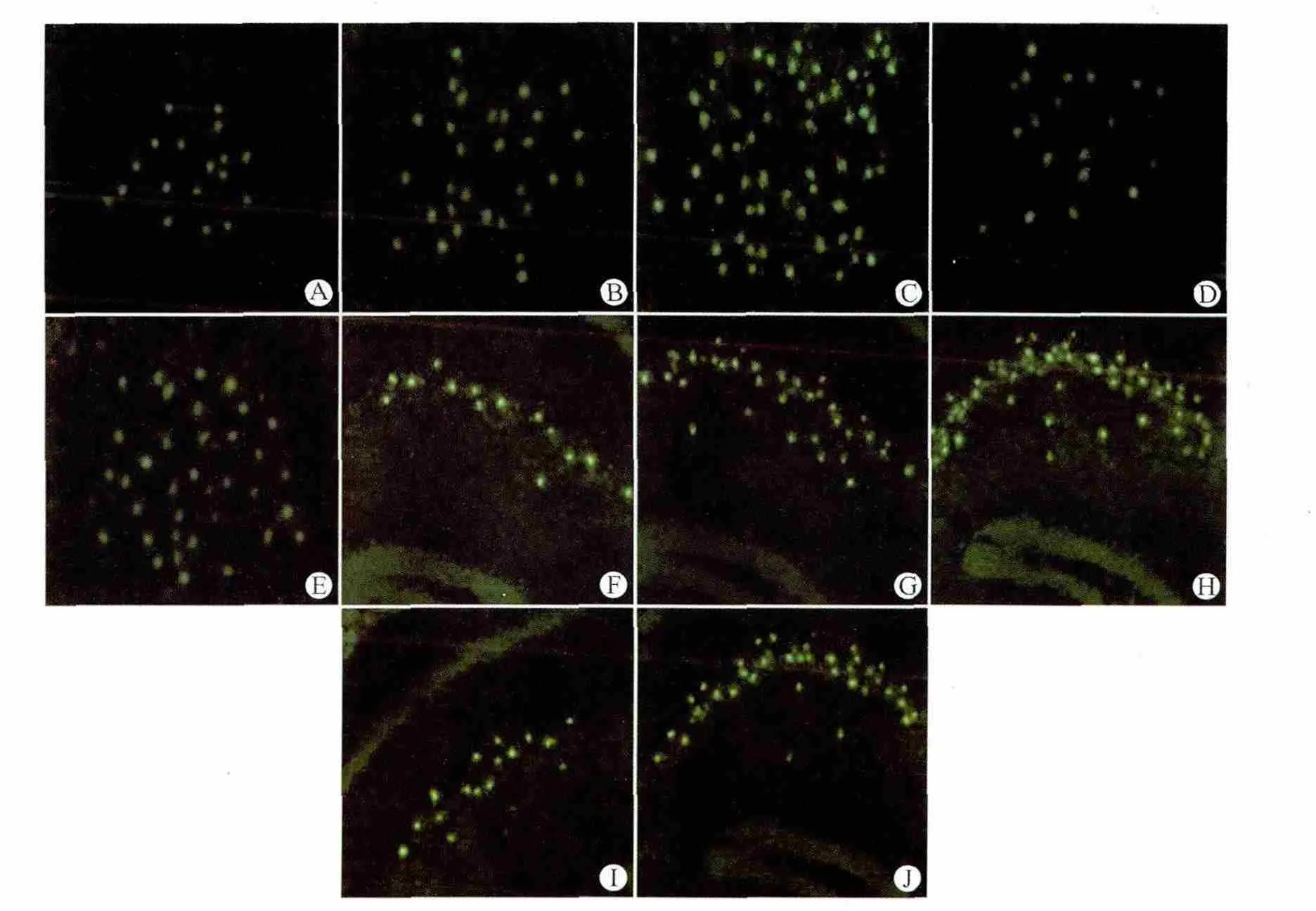
图4 各组小鼠脑内皮层和海马COX-2蛋白阳性细胞检测结果(免疫荧光,×400)Fig.4 COX-2positive cells in the cortex and hippocampus of mice in each group(immunofluorescence,×400)
3 讨论
近年大量研究表明,COX-2和PGs在癫痫发生发展的过程中发挥着重要作用。例如在海马点燃模型、红藻酸(Kainic acid,KA)诱导及基因易感El鼠的癫痫发作模型中COX-2的表达增高[7]。孙艺平等[8]亦发现匹罗卡品(30mg/kg)诱导癫痫发作大鼠海马神经元的胞质和胞核内COX-2蛋白产物表达增多,提示癫痫发作后可引起炎性因子COX-2的高表达。Joseph等[9]发现在KA诱导癫痫持续状态后2、6、24h边缘系统脑区COX-2蛋白的表达增强。COX-2与癫痫发作存在密切的关系,COX抑制剂是否对控制癫痫发作有帮助?癫痫发作引起的神经元表达COX-2持续时间短暂。但COX-2抑制剂可明显减少癫痫发作引起的神经元损伤。Dhir等[10]研究发现抑制COX活性药物如吲哚美辛或选择性COX-2抑制剂在几种癫痫模型中可降低动物癫痫发作的频率和海马细胞死亡。选择性COX-2抑制剂可延长致痫药物和电惊厥诱导产生的癫痫发作潜伏时间,降低癫痫发作持续时间和死亡率,抑制海马神经元的死亡,保护海马神经元,调节GABA能神经元,改善癫痫大鼠空间活动能力[11]。Jayaraman等[12]研究发现PTZ点燃小鼠口服10mg/kg依他昔布可使PTZ诱导惊厥发作的死亡率下降,且无神经毒副作用,对运动协调性亦无不良影响。依他昔布还可显著增加安定的血浆浓度。本研究发现塞来昔布可减轻腺苷A1受体敲除鼠癫痫发作程度以及PTZ点燃过程中的脑组织形态结构损伤,为COX-2抑制剂的应用提供了实验基础。
近年来,不少学者认为癫痫耐药性与多药转运体(如PGP、MRP1等)的高表达有关,多药转运体通过减少抗癫痫药物在脑内的水平,从而导致癫痫耐药性的产生[13-14]。国外研究报道COX抑制剂有调控多药转运体的作用,为耐药性癫痫的治疗提出了一个新的方向。Schlichtiger等[15]发现耐药性颞叶癫痫大鼠使用塞来昔布治疗6d后其自发周期性癫痫发作的频率显著减少,对镇静催眠剂的敏感性增加,认为COX-2抑制剂的预治疗可恢复镇静催眠剂在耐药性癫痫大鼠中的抗惊厥作用,为转运体介导的耐药性癫痫的治疗提供新的治疗方向。Hartz等[16]指出癫痫发作时通过N-甲基-D-天冬氨酸(NMDA)受体和COX-2使谷氨酸盐释放从而增加PGP。认为NMDA受体和COX-2可作为与血脑屏障多药耐药转运体相关的耐药性癫痫潜在的治疗靶点。van Vliet等[17]使用高选择性的 COX-2抑制剂SC-58236和NS-398能阻碍癫痫持续状态引起的PGP在海马旁皮层和腹侧海马部位的表达增加,且发现反复发作癫痫大鼠在使用COX-2抑制剂后苯妥英钠在脑中的浓度显著增加。Zibell等[18]研究发现,高选择性COX-2抑制剂NS-398和吲哚美辛可阻碍谷氨酸诱导的PGP表达增加。与此结果一致,COX-2抑制剂塞来昔布也能阻止癫痫发作诱导的大鼠脑毛细血管PGP的表达上调。以上研究均提示以慢性癫痫脑组织中COX-2为靶点来控制PGP等多药转运体表达水平是一个十分有前途的治疗策略。本研究发现,干预组和非干预组小鼠大脑皮层和海马PGP、COX-2的表达在点燃后30d时显著高于24h时,说明PTZ点燃后COX-2的表达与PGP同步,随时间延长而增高,表明COX-2可能与癫痫耐药性的产生有着密切关系。PTZ点燃后24 h时,非干预组小鼠大脑皮层和海马PGP、COX-2表达显著高于对照组,而干预组与对照组比较差异无统计学意义,结果显示接受了塞来昔布预治疗的腺苷A1受体敲除小鼠癫痫发作早期PGP、COX-2表达与癫痫发作前无明显变化,说明在癫痫发作早期COX-2抑制剂可能通过抑制COX-2炎症反应通路完全逆转腺苷功能紊乱所致癫痫耐药性的产生。PTZ点燃后同一时间点(点燃后24h、点燃后30d)干预组PGP、COX-2的表达均低于非干预组,可见塞来昔布可下调腺苷A1受体敲除鼠PTZ点燃后PGP的表达,说明COX-2介导的癫痫耐药机制很可能与腺苷系统的参与有关。这一结果与Akula等[19]的研究结果一致。其研究发现,在静脉注射PTZ之前腹腔注射4mg/kg罗非昔布能提高癫痫各阶段痫性发作阈值。亚有效剂量的罗非昔布预治疗(30mg/kg,腹腔注射)可提高亚保护剂量的腺苷(25mg/kg,腹腔注射)或2-氯腺苷(1或2mg/kg,腹腔注射)在提高痫性发作阈值方面的作用。相反,咖啡因(100或200mg/kg,腹腔注射)或茶碱(50或100mg/kg,腹腔注射),二者为非选择性腺苷A1/A2受体拮抗剂,可逆转罗非昔布的抗惊厥作用。双嘧达莫(一种腺苷摄取抑制剂,5mg/kg,腹腔注射),表现出类似罗非昔布(1mg/kg,腹腔注射)的抗惊厥作用。这项研究阐明罗非昔布提高小鼠静脉注射PTZ诱导痫性发作阈值,从而达到的抗惊厥效果很可能有腺苷能系统的参与。本实验研究及Akula等的研究提示:同时干预腺苷系统及COX-2炎性反应通路可能成为难治性癫痫新的治疗方向。
[1] JärveläJ T,Lopez-Picon F R,Holopainen I E.Age-dependent cyclooxygenase-2induction and neuronal damage after status epilepticus in the postnatal rat hippocampus[J].Epilepsia,2008,49(5):832-841.
[2] Okada K,Yuhi T,Tsuji S,et al.Cyclooxygenase-2expression in the hippocampus of genetically epilepsy susceptible E1mice was increased after seizure[J].Brain Res,2001,894(2):332-335.
[3] Schlichtiger J,Pekcec A,Bartmann H,et al.Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy[J].Br J Pharmacol,2010,160(5):1062-1071.
[4] Yang H,Chen C.Cyclooxygenase-2in synaptic signaling[J].Curr Pharm Des,2008,14(14):1443-1451.
[5] Oliveira M S,Furian A F,Rambo L M,et al.Modulation of pentylenetetrazol-induced seizures by prostaglandin E2receptors[J].Neuroscience,2008,152(4):1110-1118.
[6] Minghetti L,Greco A,Potenza R L,et al.Effects of the adenosine A2Areceptor antagonist SCH 58621on cyclooxygenase-2expression,glial activation,and brain-derived neurotrophic factor availability in a rat model of striatal neurodegeneration[J].J Neuropathl Exp Neurol,2007,66(5):366-371.
[7] Kawaguchi K,Hickey R W,Rose M E,et al.Cyclooxygenase-2expression is induced in rat brain after kainite-induced seizures and promotes neuronal death in CA3hippocampus[J].Brain Res,2005,1050(1):130-137.
[8] 孙艺平,赵永顺,于德钦,等.匹罗卡品致痫大鼠脑内NF-κB及COX-2的表达[J].神经解剖学杂志,2008,24(4):421-424.
[9] Joseph S A,Lynd-Balta E,O’Banion M K,et al.Enhanced cyclooxygenase-2expression in olfactory-limbic forebrain following kainate-induced seizures[J].Neuroscience,2006,140(3):1051-1065.
[10] Dhir A,Naidu P S,Kulkarni S K.Effect of cyclooxygenase-2(COX-2)inhibitors in various animal models(bicuculline,picrotoxin,maximal electroshock-induced convulsions)of epilepsy with possible mechanism of action[J].Indian J Exp Biol,2006,44(4):286-291.
[11] Gobbo O L,O’Mara S M.Post-treatment,but not pre-treatment,with the selective cycooxygenase-2inhibitor celecoxib markedly enhances functional recovery from kainic acid-induced neurodegeneration[J].Nueroscience,2004,125(4):317-327.
[12] Jayaraman R,Manisenthil K T,Anitha T,et al.Influence of etoricoxib on anticonvulsant activity of phenytoin and diaze-pam in experimental seizure models in mice[J].J Pharm Pharmacol,2010,62(5):610-614.
[13] 赵元淑,罗淼珊,邓镇 ,等.GAD65基因修饰的间充质干细胞在癫痫大鼠脑内的表达及作用[J].华中科技大学学报:医学版,2013,42(6):642-646.
[14] 刘志广,康慧聪,王媛,等.腺苷A1受体基因敲除小鼠戊四氮致痫后大脑皮层蛋白组学的研究[J].华中科技大学学报:医学版,2012,41(1):16-21.
[15] Schlichtiger J,Pekcec A,Bartmann H,et al.Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy[J].Br J Pharmacol,2010,160(5):1062-1071.
[16] Hartz A M,Notenboom S,Bauer B.Signaling to P-glycoprotein-A new therapeutic target to treat drug-resistant epilepsy?[J].Drug News Perspect,2009,22(7):393-397.
[17] van Vliet E A,Zibell G,Pekcec A,et al.COX-2inhibition controls P-glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats[J].Neuropharmacology,2010,58(2):404-412.
[18] Zibell G,Unkrüer B,Pekcec A,et al.Prevention of seizure-induced up-regulation of endothelial P-glycoprotein by COX-2 inhibition[J].Neuropharmacology,2009,56(5):849-855.
[19] Akula K K,Dhir A,Kulkarni S K.Rofecoxib,a selective cyclooxygenase-2(COX-2)inhibitor increases pentylenetetrazol seizure threshold in mice:possible involvement of adenosinergic mechanism[J].Epilepsy Res,2008,78(1):60-70.
猜你喜欢
检察风云(2022年5期)2022-04-05
中医眼耳鼻喉杂志(2021年2期)2021-07-21
中国医学影像技术(2020年11期)2021-01-04
中国现代医药杂志(2020年3期)2020-05-08
中国中医基础医学杂志(2020年1期)2020-03-03
心电与循环(2020年1期)2020-02-27
中国医学影像技术(2019年10期)2019-10-24
中国生物医学工程学报(2019年6期)2019-07-16
中外医疗(2016年15期)2016-12-01
国外医药(抗生素分册)(2016年3期)2016-07-12
