
儿童早发型腓骨肌萎缩症2型21例线粒体融合蛋白2基因型和表型分析
2013-12-26 02:00吕俊兰张礼萍李久伟郎志奇丁昌红陈春红
中国循证儿科杂志 2013年2期
吕俊兰 刘 京 张礼萍 李久伟 孙 欣 郎志奇 丁昌红 陈春红
·论著·
儿童早发型腓骨肌萎缩症2型21例线粒体融合蛋白2基因型和表型分析
吕俊兰1刘 京2张礼萍3李久伟1孙 欣1郎志奇4丁昌红1陈春红1
目的 分析儿童腓骨肌萎缩症2型(CMT2)线粒体融合蛋白2(MFN2 )基因型与临床表型。方法 纳入1998至2012年首都医科大学附属北京儿童医院临床诊断的CMT2患儿为研究对象,采用直接测序方法检测MFN2 突变基因,并描述分析突变和未突变患儿的临床表型、神经电生理、实验室和病理学检查特征。结果 21例CMT2患儿进入分析。①未检出MFN2 基因突变18例(男14例,女4例),起病年龄平均3.3岁。其中9例累及下肢近端和远端,7例累及四肢,2例无法行走,8例下肢近端和远端肌肉萎缩,4例可见轻微感觉障碍。MFN2 基因突变3例(男2例,女1例),起病年龄1.5~8岁,1例有家族史。均可见下肢近端和远端肌肉萎缩,2例足下垂并内翻,均有感觉障碍表现。②21例运动和(或)感觉神经传导速度≥38 m·s-1或波幅降低,MFN2 基因突变患儿腓总神经和胫神经复合肌肉动作电位波幅较正常值的下降幅度高于未突变患儿;③10例行腓肠神经活检检查,其中MFN2 基因突变1例,未检出MFN2 突变9例,均符合慢性轴索神经病病理改变, 电镜下可见轴索线粒体聚集排列和肿胀。结论MFN2 基因突变是CMT2的致病原因之一,携带该突变患儿多为早发型,临床表型可能重于未突变者,仍需扩大样本量进一步分析。
腓骨肌萎缩症2型; 线粒体融合蛋白2基因; 临床表型; 儿童
腓骨肌萎缩症(CMT) 是遗传性感觉运动神经病(HMSN)的一个最重要的亚型,临床表现为慢性进行性的多发性周围神经病。CMT依据病理和神经电生理改变可以分为2个主要亚型,脱髓鞘型(CMT1),神经传导速度<38 m·s-1;轴索型(CMT2),神经传导速度≥38 m·s-1。CMT遗传方式多样,可表现为常染色体显性、隐性或X性联遗传[1,2]。 线粒体融合蛋白2(MFN2 )基因突变可导致线粒体GTP酶的动力学障碍,是常染色体显性遗传CMT2患者的致病原因之一。国内关于CMT2患儿的报道较少,且鲜见关于其基因型和临床表型的分析。本研究前期已对CMT患儿的电生理与病理特征进行了研究[3,4],之后诊断病例逐渐增多,本文主要报道CMT2型患儿MFN2 基因突变与临床表型相关性,以期加强对该病的认识。
1 方法
1.1 纳入标准 ①首都医科大学附属北京儿童医院临床诊断CMT2的连续病例,符合Dyck周围神经病学的相关诊断标准[1]:a.以进行性对称性肌无力和萎缩为主要临床特征。足和腿首先受累,之后累及手和前臂。踝关节背屈、跖屈力量减弱,背屈减弱更为明显,伴上肢远端不同程度的肌无力。可有弓形足或锤状趾和局限性的感觉障碍, 但在体表不能触及粗大的神经;b.神经传导检查:上肢运动神经传导速度(MCV)正常(≥38 m·s-1),可有下肢MCV轻微减慢,以复合肌动作电位降低为主要特征。针刺肌电图检查表现为远端肌肉运动单位电位时限增宽、波幅增高,募集减少,伴有失神经表现;②进行MFN2 基因突变筛查者。
1.2 对照组 选取非神经系统疾病的正常儿童作为MFN2 基因检测对照,样本量与CMT2患儿1∶1匹配。
1.3MFN2 基因突变检测方法 采集静脉血提取外周血白细胞DNA。MFN2 基因的编码外显子3~19号的引物序列通过软件Primer 5.0 设计, 由上海生物工程技术服务有限公司合成; 使用ABI3730XL测序仪(美国), 采用直接测序的方法对MFN2 基因的17个外显子进行测序,应用DNA star,Chromas软件和http://blast.ncbi.nlm.nih.gov分析测序结果对比,经鉴定出的突变行反向测序验证。
1.4 神经电生理检查方法 采用KEYPOINT肌电图诱发电位仪, 检测方法及判别方法参见文献[1]。检测神经包括正中神经、尺神经、腓总神经、胫神经和腓肠神经。≥3岁神经传导速度和复合肌肉动作电位(CMAP)波幅的正常范围以健康成人正常值作为参照;<3岁为成人正常值的80%,远端潜伏期为成人正常值的120%。
1.5 腓肠神经活检病理检查 取外踝和跟腱正中切口,切取腓肠神经约2 cm,分成2段。一段中性福尔马林固定,石蜡包埋,取标本横向、纵向切片,行苏木精-伊红和刚果红(LHE)染色,光镜观察。另一段以树脂包埋,半薄切片,甲苯胺蓝染色,光镜观察。选择病变明显部位进行定位修块,超薄切片,电镜观察。由专业技术员按标准化方法完成。
1.6 分组和资料截取 依据MFN2 基因检测结果分为突变组和未突变组。并从病史中截取以下信息进行两组的描述性分析:①一般情况:性别、起病年龄、就诊年龄和病程;②临床表现:肌无力、萎缩,足和手畸形,感觉障碍等;③神经电生理检查:MCV和感觉神经传导速度(SCV);④影像学检查:头颅CT或MRI;⑤实验室检查:肌酶、CSF生化;⑥肌肉病理学检查等。
2 结果
2.1 一般情况 1998至2012 年符合CMT2临床诊断21例患儿进入分析,男16例,女5例。对照组纳入21例,平均年龄3.5岁。
2.2MFN2基因突变检测结果 21例CMT2患儿行MFN2 基因检测,发现3种突变位点,均为杂合错义突变。例5第12号外显子出现c. 1 253 G→A改变,导致MFN2蛋白第418位精氨酸被谷氨酰胺替换(R418Q)(图1A)。例8 测序结果第19号外显子出现c. 2 213 C→T改变,导致MFN2蛋白第738位丙氨酸被缬氨酸替换(A738V)(图1B)。例12测序结果第11号外显子出现c. 1 040 C→T,导致MFN2蛋白第364位精氨酸被色氨酸替换(R364W)(图1C)。查阅突变数据库和多态数据库,位于第11号外显子c.1 040 C→T,第12号外显子c. 1 253 G→A为已报道的致病热点突变;第19号外显子的c. 2 213 C→T为首次报道的杂合错义突变。21例正常对照组均没有发现上述3种基因突变。
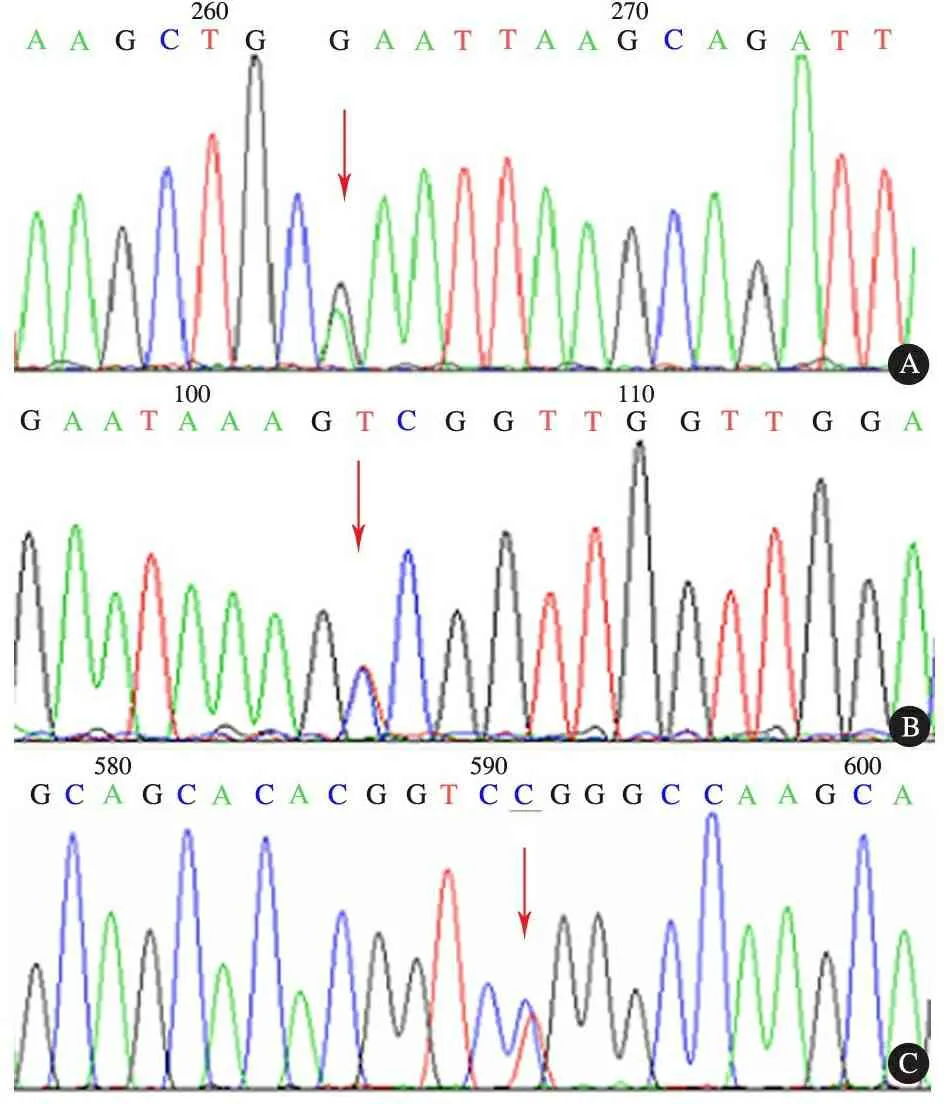
图1 本文3例患儿MFN2基因突变患儿外显子测序图
Fig 1 Mutations ofMFN2 gene in three CMT2 patients
Notes A: case 5, exon-12, c. 1253 G→A(R418Q); B: case 8, exon-9,c. 2213 C→T(A738V);C: case 12, exon-9, c. 1040 C→T(R364W)
2.3 临床表现 如表1所示,3例MFN2 基因突变患儿,男2例,女1例。起病年龄1.5~8岁。1例为常染色体显性遗传。3例均可见四肢肌肉无力和以下肢近端和远端肌肉为主的萎缩,2例足下垂并内翻。2例轻微感觉障碍,1例明显感觉障碍。
18例未检出MFN2基因突变患儿,男14例,女4例。起病年龄平均3.3岁,就诊年龄平均7.6岁,病程平均2.8岁。2例有家族史。18例均有肌肉无力表现,其中9例累及下肢近端和远端,7例累及四肢,2例无法行走;18例均可见肌肉萎缩,其中8例累及下肢近端和远端,10例累及四肢。单纯足下垂8例,足下垂并内翻9例,四肢畸形1例。4例可见轻微感觉障碍。
2.4 神经电生理检查 表2显示,除例17未行神经电生理检测外,20例正中神经MCV均>38 m·s-1, 均有运动和(或)感觉神经传导速度波幅降低(或未引出电位)。MFN2基因突变患儿腓总神经CMAP波幅平均为0.007 mV,较正常值下降97%;胫神经CMAP波幅平均为0.57 mV,较正常值下降72%;MFN2基因无突变患儿:腓总神经CMAP波幅平均为1.80 mV, 较正常值下降10%;胫神经CMAP波幅平均为1.60 mV,较正常值下降20%。非突变组, 腓总神经CMAP波幅均值为1.8, 较正常均值下降10%, 胫神经CMAP波幅均值1.6, 较正常均值下降20%。
2.5 影像学检查 5/21例行头颅CT或MRI检查,其中MFN2基因突变和未突变分别为2例和3例,1例发现有鞍上池囊肿(MFN2基因未突变,例4), 余4例正常。
2.6 实验室检查 4/21例(MFN2基因未突变3例,突变1例)血生化检查提示有肌酸激酶的轻度升高;MFN2基因突变1例(例5)CSF蛋白轻度升高(0.41 g·L-1)。
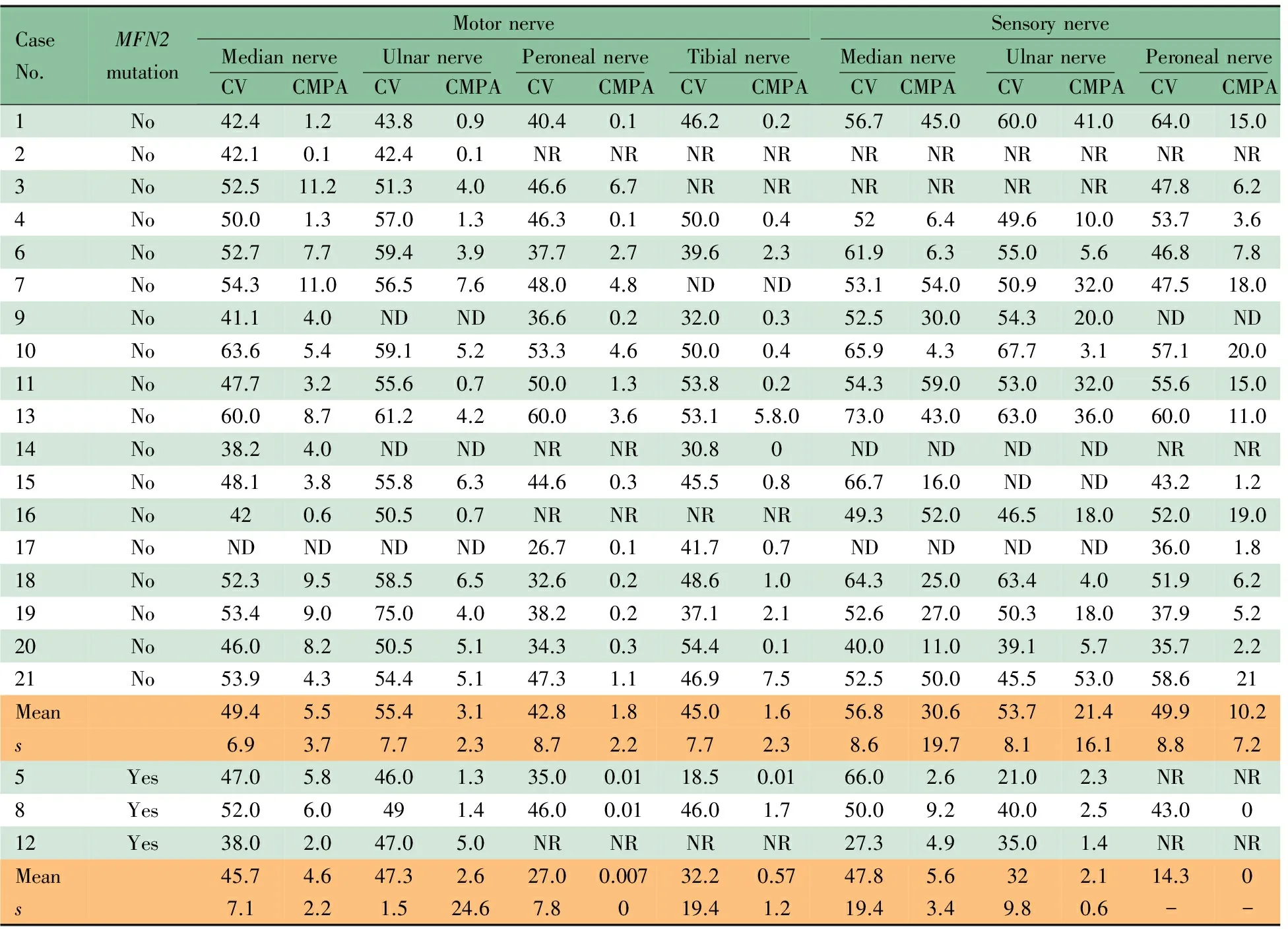
表2 MFN2基因突变和未突变患儿电生理检查结果
Notes CV: conduction velocity(m·s-1);CMPA:(mV); ND: not done; NR: not run
2.7 病理学检查 10例行腓肠神经活检,其中MFN2基因突变1例,未检出MFN2J基因突变9例,均符合慢性轴索神经病病理改变,表现为有髓神经密度的减少,轴索变性的神经纤维和再生的神经纤维束(图2A~C)。电镜检查可见轴索内聚集排列和肥大的异常线粒体(图2D)。
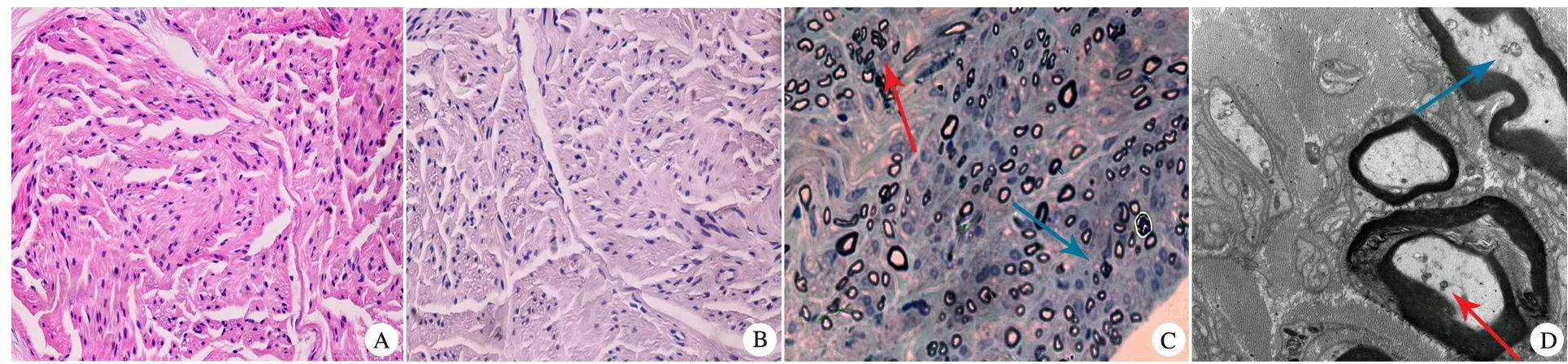
图2 腓肠神经活检光镜和电镜所见
Fig 2 Microscopic findings of sural nerve biopsy by both light and electro-microscopy
Notes A:Paraffin section,HE staining light microscope; medllated fibers were decreased, no inflammatory cell infiltration was seen.B:Frozen section,Congo red staining,no abnormal staining was observed.C :Semi-film section, methylaniline blue staining,nerve fibers decreased obviously, especially the nerve fibers with big diameter, the nerve fibers with axonal degeneration(red arrow) and regenerative nerve fibers(blue arrow) could be observed. D :Aggregation and edema of axonal mitochondria in meduated(blue arrow) and unmeduated(red arrow) nerve fibers were observed
3 讨论
MFN2是一个大的跨膜GTP酶, 位于线粒体外膜上,定位在染色体1p36.22, 其功能为促进线粒体的融合。近年来的研究表明约1/3的CMT2患者是由MFN2基因的突变所致。MFN2基因的突变可以使神经轴索变性和凋亡,导致周围神经病变。由于突变的外显子区位于或邻近GTP酶的功能区,故MFN2基因突变可通过影响细胞的氧化磷酸化作用,影响其能量代谢导致线粒体能量代谢异常,进而引起神经功能的异常[5,6]。MFN2基因已发现多种突变类型, 其中约90%为错义突变, 少数为终止突变, 尚未发现重复突变[7]。MFN2基因突变位点主要集中在GTP酶结合域(>50%),Baloh 等[8]研究发现MFN2基因突变可导致神经原轴突和感觉神经末梢线粒体运输传导能力的减弱。本文3例患儿检出的均为错义突变,且其中1个为未报道的新突变。MFN2的基因突变率与种族相关,不同国家报道的突变率有所不同,如欧美的较大样本(323例)CMT2 的研究表明MFN2基因突变率为8%[9],而来自韩国的一项研究表明突变率高达33%[10],本文CMT2患儿MFN2基因突变的检出率为14.2%(3/21),但因样本量的限制,故基因突变率难有代表意义,应进一步扩大样本量进行研究。另外, 种族的差异也是导致阳性率偏低的另一原因。本研究仅针对先证者进行了基因检测,尚未对患儿家庭成员进行深入的研究。
CMT2可分为早发型(起病年龄<10岁)和晚发型(起病年龄≥10岁)[5,10]。本文病例发病年龄最小为6个月, 最大为13岁。3例MFN2基因突变病例中发病年龄均在10岁以内,属于早发型CMT2病例。据国外的研究资料表明,MFN2基因突变相关的CMT2 也以早发型为多见[11,12]。CMT2多为常染色体隐性遗传,在MFN2基因相关的神经病变,病情的严重程度并不一致。从轻微受累到罹患严重的周围神经病均可存在,约25% 的MFN2基因突变者随着病情的进展,而严重影响其生活质量,甚至需要轮椅。但也有病情轻者,仅有肢体远端的轻度无力,甚至有报道MFN2基因突变的个体可以完全无症状[13],提示MFN2 基因突变病例具有明显的临床异质性。本文MFN2基因突变病例的肌肉无力、萎缩,足和手畸形,感觉障碍的严重程度似乎高于无突变者;同时神经传导检查显示,MFN2基因突变病例的腓总和胫神经的CAMP下降较无突变组更为明显,提示轴索变性更为严重;但由于病例数较少,尚无法行统计学检验。
本文10例行腓肠神经活检,均在基因检测前完成。均有髓神经纤维密度的明显减少和有髓神经的轴索变性阳性发现。电镜检查显示,1例MFN2 基因突变和6例无突变患儿有明显的有髓神经和无髓神经纤维的线粒体结构异常,表现为线粒体的异常肥大和聚集排列。由于周围神经活检是损伤性的检查,建议腓肠神经活检应在基因检测后进行。对于MFN2基因无突变患儿,可考虑进行腓肠神经活检,应用电镜技术可清晰的观察亚细胞结构包括线粒体是否异常[5,14 ]。
综上,MFN2基因突变多为CMT2早发型,腓总和胫神经的CAMP下降较无突变者更为明显,提示轴索变性更为严重。
[1]Peter J. Dyck PKT. Peripheral neuropathy. 4rd eded. philadelphia:W.B. Sauders,2005
[2]Ryan MM, Ouvrier R. Hereditary peripheral neuropathies of childhood. Curr Opin Neurol, 2005,18(2):105-110
[3]Lv JL(吕俊兰), Wu HS, Lang ZQ, et al. Peripheral neurophysiological and neuropathological studies on Charcot-Marie-Tooth atrophy disease in children. Chin J Pediatr(中华儿科杂志), 2002,40(3):173-176
[4]Lv JL(吕俊兰). Hereditary peripheral neuropathy in children. J Appl Clin Pedaitr(实用儿科临床杂志), 2006,21(12):723-725
[5]Calvo J, Funalot B, Ouvrier RA, et al. Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch Neurol, 2009,66(12):1511-1516
[6]Ouvrier R, Grew S. Mechanisms of disease and clinical features of mutations of the gene for mitofusin 2: an important cause of hereditary peripheral neuropathy with striking clinical variability in children and adults. Dev Med Child Neurol, 2010,52(4):328-330
[7]Engelfried K, Vorgerd M, Hagedorn M, et al. Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2). BMC Med Genet, 2006,7:53
[8]Baloh RH, Schmidt RE, Pestronk A, et al. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci, 2007,27(2):422-430
[9]Verhoeven K, Claeys KG, Zuchner S, et al. MFN2 mutation distribution and genotype/phenotype correlation in charcot-Marie-Tooth type 2. Brain, 2006, 129(Pt 8):2093-2102
[10]Chung KW, Kim SB, Park KD, et al. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain, 2006,129(Pt 8):2103-2118
[11]Kijima K, Numakura C, Izumino H, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet, 2005, 116(1-2): 23-27
[12]Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004. 36(5): 449-451
[13]Lawson VH, Graham BV, Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology,2005, 65(2): 197-204
[14]Funalot B, Magdelaine C, Sturtz F, et al. Ultrastructural lesions of axonal mitochondria in patients with childhood-onset Charcot-Marie-Tooth disease due to MFN2 mutations. Bull Acad Natl Med, 2009,193(1):151-160
Analysis of mitofusin 2 genotype and phenotype in 21 children with charact-marry-tooth type Ⅱ
LVJun-lan1,LIUJing2,ZHANGLi-ping3,LIJiu-wei1,SUNXin1,LANGZhi-qi4,DINGChang-hong1,CHENChun-hong1
(1DepartmentofNeurology,BeijingChildren′sHospital,theCapitalMedicineUniversity,Beijing100045; 2DepartmentofPediatrics,PekingUniversityPeople′sHospital,Beijing100044; 3DepartmentofPediatrics,XuanwuHospital,CapitalMedicalUniversity,Beijing100053; 4DepartmentofPathology,BeijingChildren′sHospital,theCapitalMedicineUniversity,Beijing100045,China)
LV Jun-lan, E-mail:junlanlu@gmail.com
ObjectiveTo analyzeMFN2 mutations and associated phenotypes in patients with charact-marry-tooth (CMT) type Ⅱ.MethodsChildren diagnosed as CMT 2 in Beijing Children's Hospital from 1998 to 2012 were included.Direct sequencing of theMFN2 gene and clinical investigations were performed in patients withMFN2 mutations. The clinical features, electrophysiological finding, labortory test and pathology of paitients with and withoutMFN2 mutations were analyzed. ResultsTwenty one cases with CMT 2 were included in the study.①No gene mutation was detected in 18 cases (male 14 cases, female 4 cases). The average age of the onset of the disease was 3.3 years. Nine of them involved in both proximal end and distal end of lower limb, seven cases involved in limbs, two cases could not walk, eight cases were with myophagism of proximal and distal ends of lower limb, mild sensory disturbance could be detected in four cases. Three different missense mutations were identified in 3 patients (male 2 cases, female 1 case). The age of onset ranged from 1.5 to 8 years, one had a family history of autosomal dominant inheritance. Myophagism of proximal and distal ends of lower limb could be found in all cases, foot drop with strephenopodia were observed in two cases, and all with sensory disturbance. ②Conduction velocity great than 38 m·s-1of motor nerve and/or sensory nerve, or decreased amplitude could be detected in all cases. The decreased value of CAMP of common peroneal nerve and tibial nerve was higher inMFN2 gene mutation children than that in children withoutMFN2 mutation.③Sural nerve examination was performed in ten cases, one of them withMFN2 gene mutation, nine withoutMFN2 gene mutation, prominent mitochondrial abnormalities in both myelinated and unmyelinated nerve fibers were observed under electronic microscope.ConclusionsMFN2 mutations was one of the causes of CMT2 with either dominant or recessive inheritance. The age of onset of patients withMFN2 gene mutation usually was before 10 years old.
Charact-marry-tooth type Ⅱ;MFN2 gene; Phenotype; Children
北京市教育委员会科技计划面上项目:KM200910025021
1 首都医科大学附属北京儿童医院神经内科与康复中心 北京,100045;2 北京大学人民医院儿科 北京,100044;3 首都医科大学宣武医院儿科 北京,100053;4 首都医科大学附属北京儿童医院病理科电镜室 北京,100045
吕俊兰,E-mail:junlanlu@gmail.com
10.3969/j.issn.1673-5501.2013.02.010
2012-10-27
2013-01-13)
丁俊杰)
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
实用手外科杂志(2021年2期)2021-07-08
健康之家(2021年19期)2021-05-23
创伤外科杂志(2021年1期)2021-02-04
中国生殖健康(2020年4期)2021-01-18
世界最新医学信息文摘(2020年25期)2020-12-25
云南医药(2020年5期)2020-10-27
中国生殖健康(2018年4期)2018-11-06
中国老年学杂志(2017年17期)2017-09-14
中国实用神经疾病杂志(2017年2期)2017-02-22
