
ACE2 基因敲除小鼠止血带休克后肾组织ACE/AngⅡ表达的变化及其与肾损伤的关系*
2013-12-23 06:26:46王建军杨秀红王银环刘英超王小君张连元
中国病理生理杂志 2013年5期
王建军, 杨秀红, 王银环, 刘英超, 王小君, 张 田, 孙 娜, 张连元
(河北联合大学基础医学院生理教研室,河北 唐山063000)
急性肾损伤是临床上休克后常见的并发症,其机制不详。我们的前期研究发现,止血带休克(tourniquet shock,TS)后小鼠肾组织血管紧张素转化酶(angiotensin-converting enzyme,ACE)表达上调和ACE2 表达下调,同时TS 小鼠出现肾氧化应激反应增强、肾功能损伤变化[1]。ACE 可催化血管紧张素Ⅰ(angiotensinⅠ,AngⅠ)生成AngⅡ,而ACE2 作为肾素血管紧张素系统(renin-angiotensin system,RAS)的负调控分子可将AngⅡ降解为Ang(1-7)。据文献报道,ACE2 能够预防AngⅡ介导的肾氧化应激、炎症和纤维化作用[2]。因此我们设想,ACE2 基因敲除(ACE2 KO)可能通过增加肾ACE/AngⅡ表达促进动物体内氧化应激机制而加重器官损伤。为此在本实验中,我们应用ACE2 KO 小鼠制作TS 模型,观察ACE2 KO 小鼠在TS 后肾组织ACE/AngⅡ表达变化以及肾损伤的反应,旨在进一步探讨ACE2 在休克后急性肾损伤中的作用。
材 料 和 方 法
1 材料
1.1 主要试剂 蛋白提取试剂盒、蛋白浓度测定试剂盒、Western blotting 蛋白marker、X 线片ECL 发光剂、定影剂和显影剂均为碧云天试剂公司产品;ACE、ACE2 和β-actin 抗体均购自Abcam;AngⅡ酶联免疫吸附测定ELISA 试剂盒购自武汉优尔生科技股份有限公司;丙二醛(malondialdehyde,MDA)、超氧化物歧化酶(superoxide dismutase,SOD)、血尿素氮(blood urea nitrogen,BUN)和肌酐(creatinine,Cr)测定试剂盒购自南京建成生物工程研究所。
1.2 动物 6 月龄雄性野生型和ACE2 基因敲除C57BL/6 小鼠由中国医学科学院实验动物研究所引进,于河北联合大学实验动物中心饲养,自由进食、饮水。
2 方法
按照蛋白提取试剂盒说明进行操作。将一侧肾组织100 mg 放入玻璃匀浆器,加入1 mL 蛋白裂解液匀浆,12 000 r/min 离心5 min,留取上清。用BCA 法测定蛋白浓度,用裂解液将各蛋白配成浓度为6 g/L,取10 μL 蛋白样品与5 ×上样缓冲液混匀,煮沸5 min 后SDS-PAGE 电泳,转硝酸纤维素膜3 ~5 h,5%BSA 室温封闭1 h,I 抗孵育1 h,TBST 洗膜3 次,每次5 min,Ⅱ抗孵育0.5 h,TBST 洗膜3 次,每次5 min。ECL 发光试剂均匀铺于膜上孵育2 min,曝光后,显影、定影。结果用ImageJ 图像分析软件进行分析。
2.4 ELISA 测定肾组织AngⅡ水平 将100 mg 肾组织放入玻璃匀浆器,加入5 mL PBS 研磨,12 000 r/min离心5 min,留取上清。应用竞争抑制酶联免疫分析法测定样本中AngⅡ水平。简单地说,往已包被鼠AngⅡ单克隆抗体的微孔中同时加入生物素标记的抗原和待测样品50 μL,37 ℃温育1 h。洗涤3次,然后加入HRP 标记的亲和素,37 ℃温育30 min。洗涤5 次,加入90 μL 底物TMB 显色15 min。加终止液终止反应。用酶标仪在450 nm 波长下测定吸光度,同时绘制标准曲线,计算样品浓度。
2.5 SOD 活性和MDA 含量测定 取肾组织100 mg,用RIPA 裂解液制成10%组织匀浆,离心、提取上清。硫代巴比妥酸(TBA)法测组织MDA,结果以μmol/g 蛋白表示;氮蓝四唑(NBT)法测组织SOD 活性,结果以103U/g 蛋白表示。按照试剂盒说明进行操作,用酶标仪分别在535 nm 和550 nm 波长下测定吸光度。
2.6 BUN 和血清Cr 的测定 待全血凝固后,3 000 r/min 离心5 min 取上清。二乙酰-肟比色法测定BUN 含量,结果以mmol/L 表示;苦味酸法测定血清Cr,结果以μmol/L 表示。按照试剂盒说明进行操作,用酶标仪分别在640 nm 和510 nm 波长下测定吸光度。
2.1 模型复制 采用我室常规复制小鼠TS 模型,水合氯醛按3 mL/kg 腹腔注射麻醉小鼠,用橡皮带环绕结扎小鼠双后肢根部,阻断血流2 h 后松开橡皮带使血流再灌注,于4 h 摘眼球放血处死小鼠,取血浆和肾组织,-70 ℃冰箱保存待各指标测定。
2.2 动物分组 将2 种小鼠分别随机分为(1)野生型小鼠(WT)组,不进行结扎处理;(2)野生型止血带休克(WT+TS)组,结扎止血带;(3)ACE2 基因敲除(KO)组,不进行结扎处理;(4)ACE2 基因敲除止血带休克(KO+TS)组,结扎止血带。每组6 只动物。
2.3 Western blotting 测定肾组织ACE 蛋白表达
3 统计学处理
数据以均数±标准差(mean ±SD)表示,多组间比较用单因素方差分析。统计处理均由SPSS 13.0统计软件完成。
结 果
1 肾组织ACE 表达的变化
Western blotting 显示,与WT 小鼠相比,WT +TS小鼠肾组织ACE 表达明显增加;KO 小鼠在肢体缺血再灌注后ACE 表达进一步增加,明显高于WT 小鼠;与WT+TS 小鼠比较,KO +TS 小鼠肾组织ACE表达也显著增加,见图1。
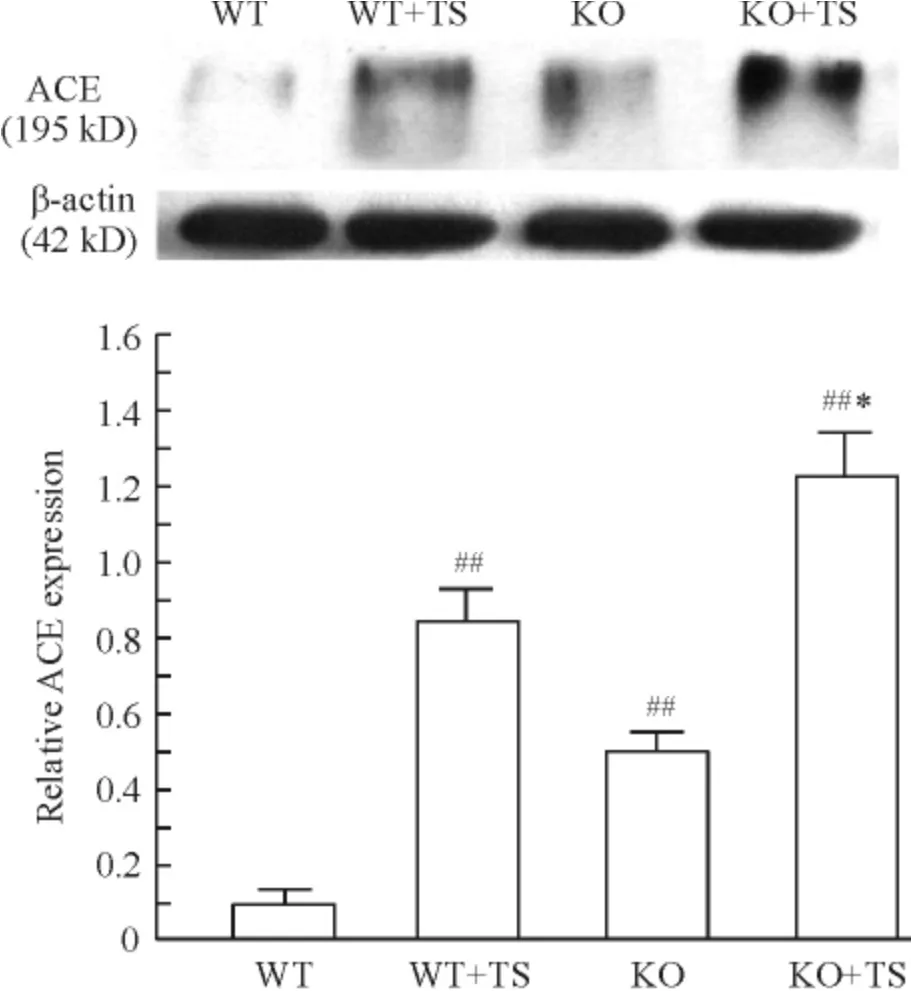
Figure 1. ACE expression in mouse kidneys of each group. Mean±SD. n=6. ##P <0.01 vs WT group;* P <0.05 vs WT+TS group.图1 各组小鼠肾组织ACE 的表达
2 肾组织AngⅡ水平的变化
ELISA 结果显示,与WT 小鼠相比,WT +TS 小鼠肾组织AngⅡ水平水平明显增加;KO 小鼠在肢体缺血再灌注后Ang Ⅱ水平进一步增加,明显高于WT小鼠;与WT + TS 小鼠比较,KO + TS 小鼠肾组织AngⅡ水平也显著增加,见图2。
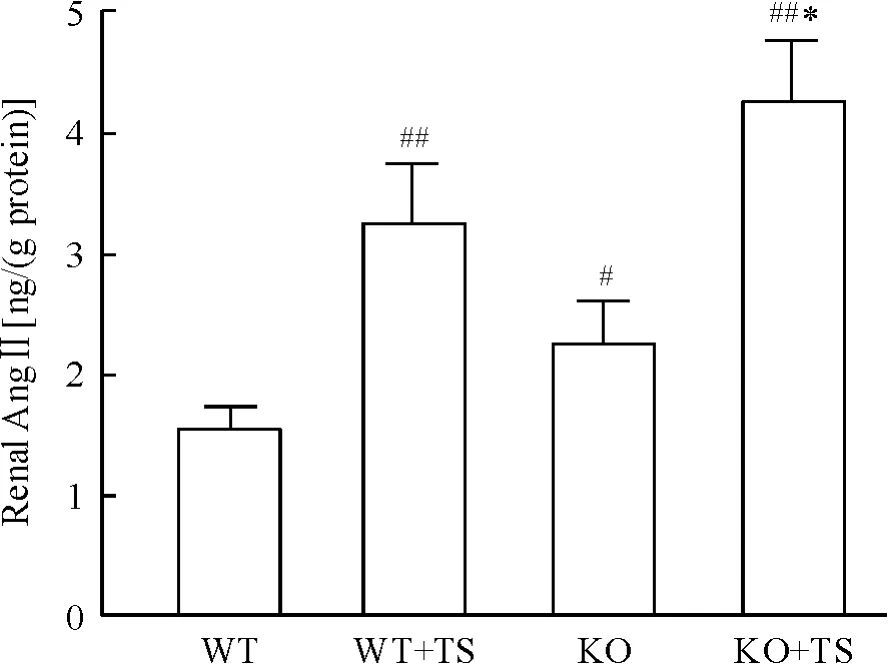
Figure 2. AngⅡlevel in mouse kidneys of each group. Mean±SD. n=6. #P <0.05,##P <0.01 vs WT group;* P<0.05 vs WT+TS group.图2 各组小鼠肾组织AngⅡ的水平
3 MDA 含量和SOD 活性测定结果
化学比色结果显示,与WT 小鼠相比,WT + TS小鼠肾组织MDA 含量明显增加;KO 小鼠肾组织MDA 含量未见明显增加,但在KO +TS 小鼠明显高于WT+TS 小鼠,见图3。与WT 小鼠相比,WT +TS小鼠肾组织SOD 活性明显降低;KO 小鼠肾组织SOD 活性无明显下降,但在KO + TS 小鼠肾组织SOD 活性明显低于WT+TS 小鼠,见图3。
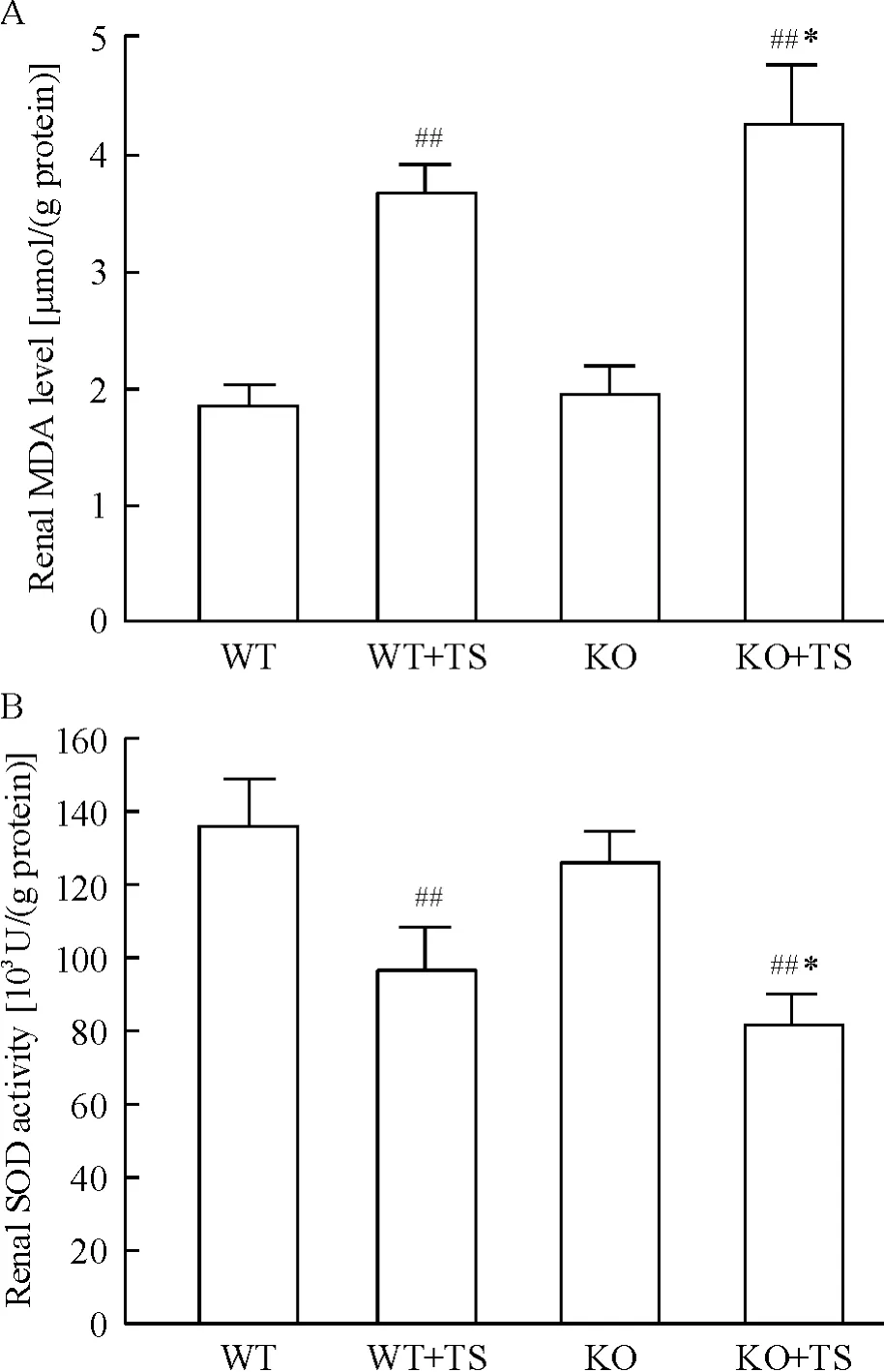
Figure 3. MDA content (A)and SOD activity (B)in kidneys of mice. Mean ± SD. n =6. ##P <0.01 vs WT group;* P <0.05 vs WT+TS group.图3 各组小鼠肾组织MDA 含量和SOD 活性
4 BUN 和血清Cr 测定结果
化学比色结果显示,WT +TS 小鼠BUN 和血清Cr 含量较WT 小鼠明显增加;与WT 小鼠相比,KO小鼠BUN 和血清Cr 含量无明显增加,但在KO +TS小鼠BUN 和血清Cr 含量均明显高于WT+TS 小鼠,见图4。
讨 论
早在几十年前人们就发现不同种类的休克与RAS 系统的激活有关,它是保证重要器官如心脏、脑等血供的一种代偿性反应。然而严重休克时RAS 系统不适当的激活对某些器官不仅不能发挥保护作用,而且还可能加重器官损伤。据文献报道[3-4],休克时局部器官如肺脏ACE 表达增加,ACE 增加能提高血清AngⅡ的水平,过量的AngⅡ通过增加氧自由基的释放加重器官损伤。尽管几十种ACE 抑制剂和血管紧张素受体阻断剂挽救和延长了许多心血管患者的生命,但在休克患者和动物的应用效果报道不一[5-6]。
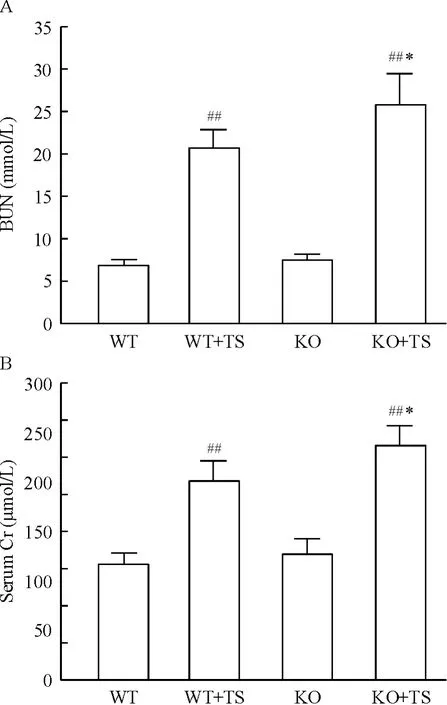
Figure 4. Content of BUN (A)and serum Cr (B)in mice of each group. Mean ± SD. n = 6. ##P <0.01 vs WT group;* P <0.05 vs WT+TS group.图4 各组小鼠TS 前后BUN 和血清Cr 的含量
目前认为RAS 还存在另外一条ACE2 催化的舒血管通路[7],2 条通路相互拮抗、相互调节,循环RAS主要发挥调节血压、维持水电平衡作用,而局部RAS在组织损伤修复中发挥主要作用。研究发现,具有抗炎作用的活性蛋白C 通过降低肾ACE 和提高肾ACE2 的表达减轻LPS 诱导的肾损伤[8];血管紧张素1 型(angiotensin type 1 receptor,AT1)受体阻断剂也可以通过抑制ACE2 的降低改善内毒素休克诱导的肺损伤[9]。这些资料表明,休克的发生不仅出现ACE 表达变化,而且负调控分子ACE2 表达变化也参与了休克的发生。
ACE 可催化AngⅠ生成AngⅡ,而ACE2 可将AngⅡ降解为Ang(1-7)。但在RAS 复杂的反应通路中,还存在其它催化途径。ACE2 不仅能降解AngⅡ,还能将AngⅠ转化成Ang(1-9);糜蛋白酶、组织蛋白酶A 也可以催化AngⅠ生成AngⅡ;除了ACE2,其它脯氨酰内肽酶、脯氨酰羧肽酶也将AngⅡ降解为Ang(1-7)[10]。因此,组织AngⅡ水平可能受多种酶催化反应的影响。我们研究发现,小鼠TS 后,肾组织ACE 表达增加,小鼠TS 后AngⅡ水平明显上升;ACE2 KO 小鼠TS 前AngⅡ水平增加,TS 后AngⅡ水平进一步增加,明显高于WT +TS 小鼠AngⅡ水平。这些结果表明,TS 时肾组织ACE 表达增加是引起AngⅡ升高的主要原因,而ACE2 基因的缺失通过降低AngⅡ分解进一步提高AngⅡ,从而影响休克的发生和进展过程。
多数学者认为,休克后多器官损伤的发生是全身炎症反应累及局部器官所致。止血带休克时大量的活性氧代谢产物从局部缺血组织释放,激活了一些内源性的炎症介质如肿瘤坏死因子α(tumor necrosis factor alpha,TNF-α)等,炎症失控导致多器官氧化应激损伤[11]。文献报道,AngⅡ的输注可明显提高肾组织TNF-α 等炎症介质的表达,而给予ACE 抑制剂可使TNF-α 的过表达消失[12];ACE2 可以抑制AngⅡ升高引起的氧化应激和炎症反应[2];ACE2 缺失促进糖尿病小鼠肾的氧化应激损伤,并与ACE 表达上调有关[13];高血压肾病的患者AngⅡ可通过AT1受体介导的信号转导机制上调ACE 并下调ACE2 的表达[14]。我们的研究发现,ACE2 基因敲除上调了小鼠ACE/AngⅡ的表达,但尚不足以引起肾的氧化应激损伤,而休克后的ACE2 基因敲除小鼠由于大量AngⅡ的产生,加重了肾的氧化应激反应和功能损伤。根据文献和我们实验结果分析表明,ACE 与ACE2 是休克诱导肾损伤中一对作用相反的重要调节因子。
[1] 王建军,杨秀红,邓 魏,等. 小鼠止血带休克后肾组织ACE/ACE2 表达变化及意义[J]. 中国病理生理杂志,2011,27(12):2399-2402.
[2] Zhong J,Guo D,Chen CB,et al. Prevention of angiotensin II-mediated renal oxidative stress,inflammation,and fibrosis by angiotensin-converting enzyme 2[J]. Hypertension,2011,57(2):314-322.
[3] Koyuncuoˇglu H,Güngör M,Hatipoˇglu I,et al. Changes in brain and lung angiotensin converting enzyme activity in various shocks[J]. Pharmacol Res Commun,1984,16(5):479-484.
[4] 张 勇,娄冬梅,李洪岩,等. 地塞米松抑制肾素-血管紧张素系统减轻大鼠急性肺损伤[J]. 中国病理生理杂志,2009,25(1):22-25.
[5] Graninger M,Marsik C,Dukic T,et al. Enalapril does not alter adhesion molecule levels in human endotoxemia[J]. Shock,2003,19(5):448-451.
[6] Schumacher J,Puchakayala MR,Binkowski K,et al.Effects of candesartan and enalaprilat on the organ-specific microvascular permeability during haemorrhagic shock in rats[J]. Br J Anaesth,2006,96(4):437-443.
[7] Muthalif MM,Benter IF,Uddin MR,et al. Signal transduction mechanism involved in angiotensin(1-7)-stimulated arachidonic acid release and prostanoid synthesis in rabbit aortic smooth cells[J]. J Pharmacol Exper Ther,1998,284(1):388-398.
[8] Gupta A,Rhodes GJ,Berg DT,et al. Activated protein C ameliorates LPS-induced acute kidney injury and downregulates renal INOS and angiotensin 2[J]. Am J Physiol Renal Physiol,2007,293(1):F245-F254.
[9] Hagiwara S,Iwasaka H,Hidaka S,et al. Antagonist of the type-1 ANG II receptor prevents against LPS-induced septic shock in rats[J]. Intensive Care Med,2009,35(8):1471-1478.
[10] Kobori H,Nangaku M,Navar LG,et al. The intrarenal renin-angiotensin system:from physiology to the pathobiology of hypertension and kidney disease[J]. Pharmacol Rev,2007,59(3):251-287.
[11] Wakai A,Wang JH,Winter DC,et al. Tourniquet-induced systemic inflammatory response in extremity surgery[J]. J Trauma,2001,51(5):922-926.
[12]Ruiz-Ortega M,Ruperez M,Lorenzo O,et al. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney[J]. Kidney Int Suppl,2002,(82):S12-S22.
[13]Wong DW,Oudit GY,Reich H,et al. Loss of angiotensin-converting enzyme-2 (Ace2)accelerates diabetic kidney injury[J]. Am J Pathol,2007,171(2):438-451.
[14]Koka V,Huang XR,Chung AC,et al. Angiotensin II upregulates angiotensin I-converting enzyme (ACE),but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway[J]. Am J Pathol,2008,172(5):1174-1183.
猜你喜欢
课堂内外·初中版(科学少年)(2023年10期)2023-12-10 19:36:37
军事文摘(2023年20期)2023-10-31 08:42:34
娃娃乐园·综合智能(2023年3期)2023-03-24 06:27:44
流行色(2021年8期)2021-11-09 11:58:44
家庭医学(下半月)(2020年4期)2020-05-30 12:42:40
现代塑料(2018年5期)2018-06-05 08:03:49
中华骨与关节外科杂志(2017年1期)2017-05-17 06:11:20
哈尔滨医药(2015年4期)2015-12-01 03:57:56
西南医科大学学报(2014年6期)2014-03-20 15:43:54
家庭医药(2012年11期)2012-04-29 00:44:03
