
高通量测序技术在宏基因组学中的应用
2013-12-01 04:47刘莉扬崔鸿飞田埂
中国医药生物技术 2013年3期
刘莉扬,崔鸿飞,田埂
随着生命科学及研究技术的不断发展,人们对生命现象的了解更加深入。微生物因为其在工业、农业、医疗卫生、环境保护等各方面的重要地位,被越来越多的研究者关注。自然状态下,微生物几乎无处不在,无论是在自然环境如土壤、海洋甚至一些极端环境(如酸矿水)中,还是在人类和动物的皮肤、口腔、肠道中,微生物都与它们所在的环境相伴相生。除生存环境极为广泛以外,微生物的数量还极为庞大,以人类为例,人类的基因总数只占人类身上微生物基因总数的1% 左右[1]。这些微生物是环境能量、物质代谢的重要中间环节和组成部分,它们有些可以代谢生成周围其他生物所必需的底物,而有些则会代谢生成毒性物质,导致环境污染,或者宿主的疾病。因此,对微生物的研究显得极为重要。
微生物的传统研究方法主要是依赖将微生物进行培养和分离(culture-dependent)。然而,到目前为止,绝大多数微生物(99% 以上)不能依靠这样的方式获得,这极大地限制了人们对微生物的研究。随着测序技术和数据处理分析能力的飞速发展,以及人们对微生物之间相互依存的共生互利和平衡关系的深入认识,一种可以对环境中所有微生物进行研究而不依赖培养的新方向——宏基因组学应运而生。
1 宏基因组简介
宏基因组(Metagenome),或称为“元基因组”,于1998年由 Handelsman等[2]在一篇研究土壤微生物的文章中首次提出,当时的定义是“微生物群落中的所有基因组的集合”。在此之后,宏基因组的概念渐渐为人们所接受,并涌现了许多针对海洋、土壤、人类肠道等微生物的典型研究工作[3-6],目前的宏基因组研究主要指对细菌的研究。
宏基因组学研究与传统微生物研究方式的最大区别在于把微生物看成一个整体,摆脱了对单个微生物培养和分离的步骤,直接对环境中所有的微生物进行研究,进而可以全面地对所有微生物进行分析。随着宏基因组学研究技术的发展和研究者兴趣的不断增加,对其研究手段和研究对象的重点也不断发生着变化,大致可以分为三个阶段:①针对 16S rRNA 为主要研究对象的核糖体 RNA 研究;②以环境中所有遗传物质为研究对象;③以环境中所有转录本为主要研究对象的宏转录组研究。狭义的宏基因组学研究指第二个阶段,本文提到的“宏基因组学”倾向于广义的概念,即三个阶段的总和。
原核生物的核糖体 RNA,尤其是 16S rRNA,由于其高度保守的序列特性,被当做可以鉴别物种的微生物系统发育的“分子钟”[7]。第一代测序读长长、准确率较高,但通量较低,比较适合对 16S rRNA 进行测序及分析。随着高通量的第二代测序(next generation sequencing,NGS)方法的诞生,由于读长较短,所以从一次测序 16S rRNA 基因全长,到只针对 16S rRNA 中的某一个或某几个高变区进行分析和研究[8-11]。
宏基因组包含着环境微生物的全部遗传信息,相比于16S rRNA 来说,宏基因组除了群落中各种微生物的分类信息以外,更包含了所有微生物的基因信息。因此,这种数据更有助于我们对群落潜在的功能进行深入分析。并且通过对基因组大小进行均一化(normalization),我们可以对群落中的微生物进行相对定量研究[12]。功能基因研究则可以通过测序序列找到特定环境下富集的功能基因[13]。宏基因组是近年研究的热点,数据量较为庞大,尤其需要高通量的测序技术和高效的数据处理能力作为依托。
宏转录组数据则包含了环境微生物的全部转录本信息。与宏基因组中研究“可能的”群落功能、代谢通路差异相比,宏转录组可以实时、实地的对微生物群落的基因表达情况进行反映[14]。在新一代测序技术出现以前,利用传统测序技术发展出了使用 EST 序列来发现新基因的方法,比较方便地得到了大量的基因序列的信息[15]。新一代测序技术的出现,给宏转录组的研究带来了新的机遇,但是由于原核生物的mRNA 较易分解、rRNA 含量极高,高质量的样本制备比较困难,因此现在的研究仍属于起步阶段[16]。
2 测序技术的发展与高通量测序技术的特点
世界第一台自动化测序仪诞生于1987年,由美国ABI公司制造,其原理基于Sanger 测序法[17]。Sanger 测序因其较长的读长(~1000 bp)和较高的测序质量(99.999%),从 20 世纪 90年代开始,就被广泛应用在生物信息学研究当中,并在人类基因组计划(human genome program,HGP)[18]中发挥了巨大的作用。但 Sanger 测序法由于测序通量太低,速度较慢,渐渐不能满足日益增多的数据需求[19]。第二代高通量测序则避免了 Sanger 测序中所需的繁琐的克隆过程,大大减少了工作量,提高了效率。随着测序技术的不断发展,单分子测序的技术,如HeliScope[20]、Picbio[21]等测序技术逐渐开始发展。但由于技术并未十分成熟,测序正确率尚有待提高,而且成本较高,单分子测序技术尚未被广泛使用。
高通量测序技术是现今应用最广泛的测序技术,其特点是成本低、通量高、速度快,可以快速产生大量的数据。高通量测序技术的读长普遍较短,目前三个应用较多的主流平台中, Roche 454 GS FLX Tianium能测 450~800 bp,Illumina HiSeq 2000能测 150 bp(单向),其新推出的MiSeq 平台最长可测至 250 bp(单向),SOLiD 5500xl能测 75 bp(单向)。它们的测序深度可以在一定程度上弥补读长较短所带来的问题,深入并且快速的测序过程也使它们得以成为现今应用最广泛的测序技术(表1)。
3 高通量测序技术在宏基因组学研究中的应用
3.1 基于16S rRNA的微生物群落分析
原核生物的16S rRNA 基因,由于其具有鉴别物种信息的作用,被广泛地应用在了微生物群落物种多样性的分析上。16S rRNA的数据库资源较为丰富,如 RDP[22]、Greengene[23]、SILVA[24]等都是一些比较成熟、不断完善并被广泛使用的数据库,并有一些自带的分类工具(比如 RDP数据库的RDP classifer等)便于分析使用。
在鉴定物种方面,两条 16S rRNA 基因的比对差异小于3%,则可以认为是同一个物种(species);差异小于5%,则可认为是同一个属(genus);差异小于10%,则可认为是同一个科(family)。通常研究者将环境微生物群落中的16S rRNA 区域通过 PCR 进行扩增和测序,并将测得的序列比对到已有的16S rRNA 数据库中,通过数据库中的海量数据,对每条 16S rRNA的分类位置进行标定,从而得到微生物群落的物种构成、各个物种的丰度等信息。此外,鉴于已知的16S rRNA 数据库中信息有限,用比对已有数据库的方法无法对未知的16S rRNA 进行估计,因此还可以将 16S rRNA 序列聚类成分类操作逻辑单元(operational taxonomic unit,OTU),利用 OTU的数目、各个 OTU的序列数来分析估计物种多样性和丰度。此外,第一代测序由于测序长度较长,所以多采用全长的16S rRNA 测序进行分析。而第二代的高通量测序,由于其读长较短,无法覆盖全长,因此许多研究都对 16S rRNA的一个或几个高变区进行测序分析。尽管不分析全长序列,由于高通量测序的覆盖深度非常高,对物种多样性的分析仍十分有利。
由于16S rRNA的分析目前已比较成熟,所以已有很多相关的研究,包括人体环境(如皮肤、口腔、肠道、女性阴道等),自然环境(土壤、海洋等)的各类环境微生物群落进行分析。2008年,美国科罗拉多大学的Fierer等[25]采集了 51个健康年轻人的手部皮肤表面的微生物样本并利用 Roche 454 GS FLX 测序仪对其 16S rRNA 进行了测序,研究了性别、用手习惯(即是否左撇子)、洗手习惯等对手表面细菌群落多样性的影响。2009年,Lazarevic等[8]采集了 3个健康成年人的口腔微生物,对其 V5 区域进行扩增并用 Illumina 进行测序,把 V5 区域当作分类标志,对人类口腔微生物群落的多样性进行了分析。同年,Turnbaugh等[9]采集了 31 对同卵双生和23 对异卵双生的双胞胎以及其母亲的粪便样本,进行肠道微生物研究,分析环境、肥胖情况等对人体肠道微生物的影响。该研究除用Sanger 测序法测了全长的16S rRNA 序列以外,还用 454 GS FLX 测序仪对 16S rRNA的V2和V6 区进行了深度测序,并以此为分类标志进行物种多样性的分析。除人体微生物的研究以外,环境微生物也是一个大的研究方向。如2007年,Roesch等[10]利用 454 GS FLX 测序技术,对来自西半球的4个土壤样本中微生物 16S rRNA的V9 高变区进行了测序,并对其生物多样性进行了分析。
值得一提的是,16S rRNA的应用也可与我国传统中医紧密联系起来。2012年,清华大学的Jiang等[11]邀请了19 位患有慢性萎缩胃炎的志愿者,并通过传统的舌苔情况,参照其症状进行判断,将志愿者分为寒症、热证,并与另外8 位健康志愿者同时进行舌苔样本的采集,用 Illumina GAIIx 测序平台对其微生物的V6 高变区进行测序,分析舌苔微生物群落与寒热症之间的关系,并认为舌苔微生物群落可以作为人体健康状态的一个标志。
高通量测序技术在基于16S rRNA的微生物群落分析中的要点在于产生测序覆盖深度极深的16S rRNA的测序数据,并通过比对或聚类的分析方法,对数据来源的微生物物种进行分析,并估计微生物群落的物种构成。相信随着高通量测序技术的发展,可测序列长度会越来越长,更多研究在分析 16S rRNA 时会选择进行全长分析,从而在微生物群落研究中得到精确的结果。
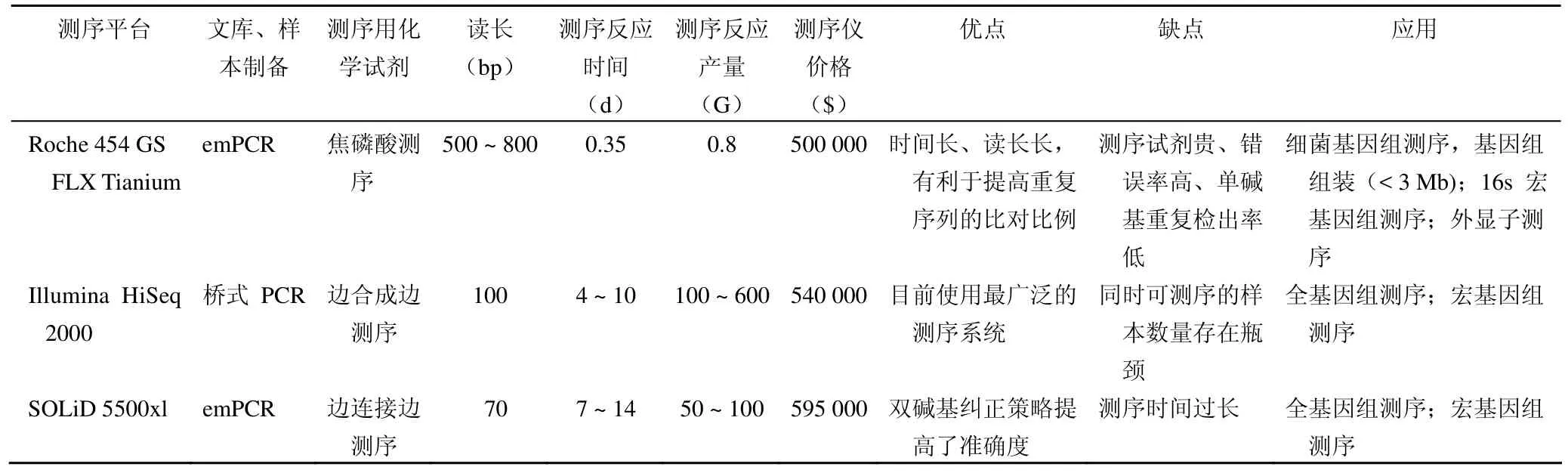
表1 三大测序平台基本情况比较
3.2 基于宏基因组的功能基因分析
对 16S rRNA的测序可以快捷地对环境微生物的群落构成进行深入的分析,除了物种多样性以外,希望得到更多的信息,比如基因信息等。在原核生物中,已知的物种只占极少的一部分,对已知物种的功能、代谢等的研究相比于未知微生物依然是微不足道的。只了解环境微生物的物种信息,远不能满足对于环境微生物群落与环境之间关系的探究,而且原核生物的变异速度很快,即使是同一个种级别内部的两个菌株在功能上都可能有非常大的区别[7]。因此研究环境微生物的全基因组就显得非常必要。
在第一代测序的条件下,由于测序速度和成本的限制,对环境内所有微生物的全基因组进行深度测序并不方便,而高通量测序则使之变成了可能。从环境微生物所有遗传信息中,可以分析和预测出该环境微生物群落可能的功能,其与环境可能的相互作用关系。
针对这种宏基因组的数据的分析,一般分为基于比对(alignment-based)的方法和不基于比对(alignment-free)的方法。基于比对的方法把测序得到的所有读段比对到已知的微生物核苷酸数据库上,如 NCBI的NT 数据库(利用Blastp等工具),或者是蛋白质 NR 数据库(利用 Blastx等工具),得到环境微生物在物种或功能基因上的丰度信息,进而结合一些功能基因、代谢通路、信号通路等数据库,对研究者感兴趣的部分进行分析。事实上,在基于比对的方法中,高通量测序所得的序列较短,而这种短序列直接进行比对的效果往往不理想[26],并且大量的原始数据进行比对会耗费很多时间,因此需要在比对前进行序列拼接,将其拼接成较长的序列,提高分析效率和分析效果。此外,还可以用一些工具对序列进行基因预测(如 Metagene[27]、GeneMark[28]、FragGeneScan[29]等)。基于比对处理高通量测序的宏基因组数据的应用非常多,2010年,华大基因在Nature 发表文章,对人体肠道微生物基因组研究计划(MetaHIT)进行了总结[30]。该研究为研究人体肠道微生物群落与人类健康之间的关系,采集了 124个欧洲人的粪便样本,其中包括 25个炎症性肠病(inflammatory bowel disease,IBD)患者和99个健康志愿者的样本,并用Illumina 测序平台进行了测序,产生了 567.7 G的测序数据,并对序列进行了拼接、注释、功能基因的分类、多态性分析等研究。2012年,华大基因在Nature 发表了一篇研究人体肠道微生物与II 型糖尿病之间关系的文章[31]。该研究收集了 345个中国人的肠道微生物样本,用 Illumina测序平台对其进行了深度测序,并在全基因组关联研究(genome wide association studies,GWAS)的基础上,开发了一种叫做全宏基因组相关联研究(metagenome wide association studies,MGWAS)的方法,对 II 型糖尿病与肠道微生物失调之间的关系进行了深入的研究。
基于比对的方法准确性较高,由于已知的数据库有限,且比对花费的时间成本非常高。所以,在基于比对的方法之外,也产生了很多不基于比对的方法和应用。不基于比对的方法大多根据序列特征,以连续 k个碱基组成的短的寡核苷酸序列(k 字词、k-mer、k-tuple)作为特征,统计这些特征在序列中出现的频数,并构建所有 4k个 k 字词的频数(频率)向量。已有研究表明这种k 字词在微生物基因组中的出现频率可以分辨微生物的不同物种[32]。基于k 字词的方法大多数被应用在快速对测序序列进行物种分类的方面(binning),这种方法的基本思想是将序列的k 字词出现频数(频率)向量与数据库中的微生物各个物种的k 字词向量作比较,将相近的划归为一组,如 AbundanceBin[33]、MetaCluster[34]等都是基于这种方法进行序列的物种划分的工具。此外 k 字词的方法也可以应用于分析样本之间的差异。如 Willner等[35]于2009年发表文章,对 86个宏基因组样本,分别用长度 k=2、3、4的k 字词进行了统计,为每个宏基因组样本构建一个 k 字词的频数(频率)向量,并对 86个样本的向量进行主成分分析、层次聚类等分析和观察。不基于比对的方法避开了复杂的计算量,在对于宏基因组的这种以未知物种为主的分析,k 字词分析的优势非常明显,将成为宏基因组的一个重要的研究方向。
高通量测序在宏基因组分析中的应用,由于分析方法的多样性,要点也不一而同。但总的来说,基于比对的方法一般需要进行序列拼接、基因预测、基因比对进而对群落的基因功能进行分析,而不基于比对的方法一般直接对序列特征进行统计。
3.3 基于宏转录组的群落转录调控规律分析
宏基因组可以详细地展示环境微生物群落中的所有遗传信息。为了精确地了解环境中正在发生的代谢过程,宏转录组的概念越来越多地被研究者们重视起来。相较于单纯的微生物基因组信息,宏转录组记录了特定时间、特定地点的微生物群落的表达谱。在活的微生物中,在某个特定时间,也并非全部基因都参与表达,而是随着环境、生长周期的变化,一部分基因有选择地被激活,进行表达。宏转录组学可以实时地记录这些活跃的基因及它们的表达量。在宏基因组中,一些已经死亡却尚未被分解的微生物的遗传信息依然可以被检测到,这些微生物本身已经不主动参与到环境的代谢当中,但是由于它们被检测到,从而对研究的结果产生一定影响。
宏转录组学的主要方法是对环境微生物样本中的mRNA 进行提取和扩增,反转录成为cDNA 并进行测序。宏转录组的实验难度较大,一方面是由于原核生物的转录和翻译同时进行,mRNA 几乎没有修饰,容易被降解,半衰期极短(约为分钟量级),因此制备高质量的样品库是实验成功的关键。另一方面,由于原核生物的rRNA 占全部RNA的比例非常大(约 70%~90%)[16,36-37],因此在制备样品时通常需要去掉 rRNA,以降低测序成本,有效地去除样本中的rRNA 也成为了一个重要课题。
宏转录组学从 2007年开始,已经有很多的相关研究,几乎所有的研究都是由高通量测序提供的数据。如 2010年,Poroyko等[38]用 454GS FLX 测序平台对两组小猪(一组为母乳喂养,另一组为配方奶喂养)的肠道微生物进行了转录组测序;2012年,Xiong等[39]对非肥胖者糖尿病(non-obese diabetic,NOD)的老鼠进行研究,设计了 8 种微生物植入无菌老鼠的肠道,培养后以不同试剂盒制备样品,并用 Illumina 平台进行转录组测序。随着实验技术的发展,已经有越来越多的宏转录组数据相继发表出来。
高通量测序在宏转录组中的应用,要点与在宏基因组分析中的应用类似。但由于技术尚在摸索之中,现阶段的难点依然在于测序前样品的制备和保存。
3.4 单细胞分离及宏基因组研究
由于宏基因组研究在组装微生物基因组和研究相似基因序列功能上的局限,当研究深入到一定的水平以后,研究者又对群体中每一个细菌的作用和不同细菌的相互关联产生兴趣。以单细胞分离、扩增为主要方法的单细胞测序方法应运而生[40-42]。单细胞宏基因组,是指将环境里所有微生物进行单个细胞的分离,而后通过全基因组扩增,或者提取RNA 反转录后进行扩增,来研究群体里单个细胞的基因组和转录组,进而得到整个群体更加完整的信息。单细胞的研究,在很多方面具有较大的优势,但在技术上还是遇到了一些问题。微生物群落巨大,分离单细胞本身就是一个非常有挑战性的工作,目前主要应用的方法,是利用流式细胞仪,将细胞通过各种染色方法进行染色,通过各种染色特性来进行区分,而细胞的染色特征可能因为不同的状态而存在差异,因此存在分离不纯的问题[42];在扩增技术上目前还没有实现突破,有扩增带来的偏向性(bias),组装基因组形成了一定的困难[41];随着单分子测序技术的不断发展,未来单分子和单细胞的结合,必将会为宏基因组研究带来新的突破。
4 结语
高通量测序技术通量高、速度快,适合宏基因组的深度测序研究。已经有相当多的宏基因组研究工作建立在高通量测序技术上,揭示微生物与环境之间的关系。同时,高通量测序读长短、数据量大的特点,对于宏基因组数据的处理也是一个挑战,催生出许多宏基因组特有的算法和工具。随着高通量测序技术的发展,为宏基因组学研究带来更多的机会。未来的高通量测序技术,除了进一步发展其通量高的优势以外,读长也会逐渐增加,同时测序错误率也会更低,现阶段研究中遇到的问题将逐步得到解决和改善。此外,目标为单分子测序的第三代测序技术的发展,也会带来全新的数据特点,宏基因组学研究将有更多的机会和发展空间。
[1]Ley RE, Peterson DA, Gordon JI.Ecological and evolutionary forces shaping microbial diversity in the human intestine.Cell, 2006, 124(4):837-848.
[2]Handelsman J, Rondon MR, Brady SF, et al.Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products.Chem Biol, 1998, 5(10):R245-R249.
[3]Dusko Ehrlich S, MetaHIT consortium.Metagenomics of the intestinal microbiota: potential applications.Gastroenterol Clin Biol,2010, 34 Suppl 1:S23-S28.
[4]Turnbaugh PJ, Ley RE, Hamady M, et al.The human microbiome project.Nature, 2007, 449(7164):804-810.
[5]Gilbert JA, Meyer F, Jansson J, et al.The Earth Microbiome Project:Meeting report of the "1 EMP meeting on sample selection and acquisition" at Argonne National Laboratory October 62010.Stand Genomic Sci, 2010, 3(3):249-253.
[6]Williamson SJ, Rusch DB, Yooseph S, et al.The Sorcerer II Global Ocean Sampling Expedition: metagenomic characterization of viruses within aquatic microbial samples.PLoS One, 2008, 3(1):e1456.
[7]Woese CR.Bacterial evolution.Microbiol Rev, 1987, 51(2):221-271.
[8]Lazarevic V, Whiteson K, Huse S, et al.Metagenomic study of the oral microbiota by Illumina high-throughput sequencing.J Microbiol Methods, 2009, 79(3):266-271.
[9]Turnbaugh PJ, Hamady M, Yatsunenko T, et al.A core gut microbiome in obese and lean twins.Nature, 2009, 457(7228):480-484.
[10]Roesch LF, Fulthorpe RR, Riva A, et al.Pyrosequencing enumerates and contrasts soil microbial diversity.ISME J, 2007, 1(4):283-290.
[11]Jiang B, Liang X, Chen Y, et al.Integrating next-generation sequencing and traditional tongue diagnosis to determine tongue coating microbiome.Sci Rep, 2012, 2:936.
[12]Frank JA, Sorensen SJ.Quantitative metagenomic analyses based on average genome size normalization.Appl Environ Microbiol, 2011,77(7):2513-2521.
[13]Allen HK, Moe LA, Rodbumrer J, et al.Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil.ISME J,2009, 3(2):243-251.
[14]Gilbert JA, Field D, Huang Y, et al.Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities.PLoS One, 2008, 3(8):e3042.
[15]Tartar A, Wheeler MM, Zhou X, et al.Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes.Biotechnol Biofuels, 2009, 2:25.
[16]Chappell L.Finding a needle in a haystack.Microbial metatranscriptomes.Nat Rev Microbiol, 2012, 10(7):446.
[17]Ner SS, Goodin DB, Pielak GJ, et al.A rapid droplet method for Sanger dideoxy sequencing.Biotechniques, 1988, 6(5):408, 410, 412.
[18]Human genome program.Science, 1989, 246(4932):873-874.
[19]Shendure J, Ji H.Next-generation DNA sequencing.Nat Biotechnol,2008, 26(10):1135-1145.
[20]Harris TD, Buzby PR, Babcock H, et al.Single-molecule DNA sequencing of a viral genome.Science, 2008, 320(5872):106-109.
[21]Eid J, Fehr A, Gray J, et al.Real-time DNA sequencing from single polymerase molecules.Science, 2009, 323(5910):133-138.
[22]Cole JR, Wang Q, Cardenas E, et al.The Ribosomal Database Project:improved alignments and new tools for rRNA analysis.Nucleic Acids Res, 2009, 37(Database issue):D141-D145.
[23]DeSantis TZ, Hugenholtz P, Larsen N, et al.Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB.Appl Environ Microbiol, 2006, 72(7):5069-5072.
[24]Quast C, Pruesse E, Yilmaz P, et al.The SILVA ribosomal RNA gene database project: improved data processing and web-based tools.Nucleic Acids Res, 2013, 41(Database issue):D590-D596.
[25]Fierer N, Hamady M, Lauber CL, et al.The influence of sex,handedness, and washing on the diversity of hand surface bacteria.Proc Natl Acad Sci U S A, 2008, 105(46):17994-17999.
[26]Prakash T, Taylor TD.Functional assignment of metagenomic data:challenges and applications.Brief Bioinform, 2012, 13(6):711-727.
[27]Noguchi H, Park J, Takagi T.MetaGene: prokaryotic gene finding from environmental genome shotgun sequences.Nucleic Acids Res,2006, 34(19):5623-5630.
[28]Zhu W, Lomsadze A, Borodovsky M.Ab initio gene identification in metagenomic sequences.Nucleic Acids Res, 2010, 38(12):e132.
[29]Rho M, Tang H, Ye Y.FragGeneScan: predicting genes in short and error-prone reads.Nucleic Acids Res, 2010, 38(20):e191.
[30]Qin J, Li R, Raes J, et al.A human gut microbial gene catalogue established by metagenomic sequencing.Nature, 2010, 464(7285):59-65.
[31]Qin J, Li Y, Cai Z, et al.A metagenome-wide association study of gut microbiota in type 2 diabetes.Nature, 2012, 490(7418):55-60.
[32]Pride DT, Meinersmann RJ, Wassenaar TM, et al.Evolutionary implications of microbial genome tetranucleotide frequency biases.Genome Res, 2003, 13(2):145-158.
[33]Wu YW, Ye Y.A novel abundance-based algorithm for binning metagenomic sequences using l-tuples.J Comput Biol, 2011, 18(3):523-534.
[34]Wang Y, Leung HC, Yiu SM, et al.MetaCluster 5.0: a two-round binning approach for metagenomic data for low-abundance species in a noisy sample.Bioinformatics, 2012, 28(18):i356-i362.
[35]Willner D, Thurber RV, Rohwer F.Metagenomic signatures of 86 microbial and viral metagenomes.Environ Microbiol, 2009, 11(7):1752-1766.
[36]Giannoukos G, Ciulla DM, Huang K, et al.Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes.Genome Biol, 2012, 13(3):R23.
[37]Schmieder R, Lim YW, Edwards R.Identification and removal of ribosomal RNA sequences from metatranscriptomes.Bioinformatics,2012, 28(3):433-435.
[38]Poroyko V, White JR, Wang M, et al.Gut microbial gene expression in mother-fed and formula-fed piglets.PLoS One, 2010, 5(8):e12459.
[39]Xiong X, Frank DN, Robertson CE, et al.Generation and analysis of a mouse intestinal metatranscriptome through Illumina based RNA-sequencing.PLoS One, 2012, 7(4):e36009.
[40]Mason OU, Hazen TC, Borglin S, et al.Metagenome,metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill.ISME J, 2012, 6(9):1715-1727.
[41]Chitsaz H, Yee-Greenbaum JL, Tesler G, et al.Efficient de novo assembly of single-cell bacterial genomes from short-read data sets.Nat biotechnol, 2011, 29(10):915-921.
[42]Stepanauskas R, Sieracki ME.Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time.Proc Natl Acad Sci U S A, 2007, 104(21):9052-9057.
猜你喜欢
当代水产(2022年8期)2022-09-20
军事文摘(2022年16期)2022-08-24
中国典型病例大全(2022年11期)2022-05-13
昆明医科大学学报(2022年2期)2022-03-29
食品安全导刊(2021年20期)2021-08-30
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
透析与人工器官(2020年1期)2020-11-16
中西医结合肝病杂志(2020年2期)2020-10-27
河南科学(2020年3期)2020-06-02
