
米哚妥林对P糖蛋白介导的多药耐药的逆转作用
2013-11-12 06:54:02魏寅祥任志广贾砚寒李新颖马远方
中国药理学与毒理学杂志 2013年1期
魏寅祥,赵 青,任志广,贾砚寒,李新颖,黎 燕,彭 晖,马远方
(1.河南大学医学院细胞与分子免疫学实验室,河南开封 475003;2.军事医学科学院基础医学研究所分子免疫室,北京 100850)
肿瘤多药耐药(multidrug resistance,MDR)指患者接受化疗药物治疗后对原来药物及其他结构和作用机制不同的抗癌药产生耐药的现象,是目前肿瘤化疗失败的主要原因。MDR的发生与多种因素有关,其中 ATP结合盒转运蛋白(ATP-binding cassette transporter,ABC转运蛋白)能量依赖性地将结构和作用机制不同的抗肿瘤药物外排到肿瘤细胞外是产生MDR的主要原因之一[1]。ABC转运蛋白超家族包括48个成员,按其序列相似性可分为ABCA到ABCG7个大类。其中P糖蛋白(P-glycoprotein,P-gp)、MDR相关蛋白(MDR associated-proteins,MRP)和乳腺癌耐药蛋白(breast cancer resistance protein,BCRP)与MDR 关系最为密切[2]。
小分子蛋白激酶抑制剂(small molecular-protein kinase inhibitor,SM-PKI)通过抑制信号通路中关键位点抑制肿瘤细胞增殖和存活,是近年来抗肿瘤药物研究的热点[3]。虽然具有靶向性强和副作用小等优点,但临床应用中出现的耐药现象依然是此类抗癌药难以回避的问题[4]。研究报道,伊马替尼诱导耐药细胞中P-gp过表达,提示P-gp可能与伊马替尼耐药具有相关性[5]。而最近越来越多的研究表明,多种SM-PKI如伊马替尼、厄洛替尼、尼洛替尼、吉非替尼、兰帕替尼、阿帕替尼和 MDRABC 间存在多种作用关系[5-9]。一方面 MDRABC会影响药物的吸收、分布、代谢和清除,而另一方面一些亲和力较高的PKI则被用于逆转相应ABC转运蛋白介导的MDR。因此,对于两者间作用关系的研究无论对于临床用药还是新药开发都具有重要的意义。
米哚妥林(midostaurin)是Novartis公司研究开发的抗肿瘤药物,为蛋白激酶C(protein kinase C,PKC)、人 FMS样酪氨酸蛋白激酶(FMS-like tyrosine kinase 3,FLT3)、KIT原癌基因蛋白,血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR),血小板衍生生长因子(plateletderived growth factor,PDGF)受体蛋白激酶抑制剂,通过阻断激酶的活化抑制肿瘤细胞的分化与增殖。米哚妥林目前已经进入Ⅲ期临床,主要应用于白血病的治疗[10],但关于米哚妥林与P-gp及MDR之间作用关系少有报道。本文用P-gp高表达的耐药细胞K562/A02及其亲本细胞K562。在体外从细胞、功能、蛋白、基因和ATP水解酶活性等多个水平,对米哚妥林与P-gp间相互作用关系进行评价,并对相关机制进行探索。
1 材料与方法
1.1 试剂和仪器
米哚妥林、罗丹明123(Rh-123)和DMSO购自美国Sigma公司;多柔比星、紫杉醇和长春新碱均购自浙江海正药业有限公司;维拉帕米购自上海禾丰制药有限公司;MTS细胞毒检测试剂盒及V3601型P-gp-GloTM检测系统购自美国Promega公司;BCA蛋白定量试剂盒购美国Thermo公司;抗P-gp单抗C219购自英国Abcam公司;AKT,pAKT(Ser473),Erk和 pErk(Thr202/Tyr204)抗体均购自美国Cell Signaling Technology公司;HRP标记的羊抗兔和羊抗鼠二抗购自美国KPL公司;Trizol购自美国Invitrogen公司;TransScript First-Stand cDNA Synthesis Supermix逆转录试剂盒自北京全式金公司;SYBR Green Realtime PCR Master Mix购自日本Toyobo公司;RPMI 1640细胞培养液购自美国Hyclone公司;胎牛血清购自元亨金马;其余试剂均为进口或国产分析纯试剂。细胞培养板购自美国Corning公司;硝酸纤维素膜购自美国Millipore公司;X线片购自美国Kodak公司。
CO2细胞培养箱(美国Thermo公司),光学倒置显微镜(日本Olympus公司),高速台式离心机(德国 Eppendorf公司),多功能酶标仪(美国Thermo公司),流式细胞仪(美国 BD公司),TD-20荧光光度计(美国PE公司),紫外分光光度计(美国Spectronic Genesys 5公司),实时定量PCR仪和 SDS-PAG电泳系统(美国 Bio-Rad公司)。
1.2 细胞培养
人慢性粒细胞白血病细胞K562由本室保存,相应的P-gp高表达耐药细胞K562/A02为多柔比星诱导得到,由中国医学科学院血液学研究所药物室熊冬生教授惠赠。两种细胞均以含10%胎牛血清的RPMI 1640培养基置于37℃及5%CO2条件恒温孵箱培养,取对数期细胞进行实验。其中K562/A02细胞的培养基中加入多柔比星1.0 mg·L-1维持其耐药性及P-gp的高表达,实验前1周停药。
1.3 MTS法检测米哚妥林细胞毒及逆转耐药作用
K562/A02与K562细胞分别以10%FBS的1640培养基悬浮,按照每180 μl 8×103个细胞接种于96孔微孔板,在5%CO2,37℃条件下培养24 h。加入米哚妥林稀释液20 μl至终浓度分别为0.2,0.5,1 和 5 μmol·L-1,于 5%CO2,37℃培养72 h。以5%DMSO组为对照,以1640培养基为本底。检测时各组按每孔10 μl加入MTS,37℃孵育3 h测定492 nm吸光度值(absorbance,A)。细胞存活率(%)=A实验组/ADMSO组×100%。
K562/A02与K562细胞分别以10%FBS的1640培养基悬浮,按照8×103接种于96孔微孔板,在5%CO2、37℃条件下培养24 h。分别按浓度梯度加入米哚妥林和维拉帕米溶液20 μl,孵育1 h后加入P-gp底物类抗癌药,并将其浓度控制在IC10左右。K562/A02中多柔比星、紫杉醇和长春新碱在 K562/A02 中相应浓度依次分别为 4 μmol·L-1,120和150 nmol·L-1,在K562中浓度为6,7.5和20 nmol·L-1。同时设置0.5‰DMSO 对照和抗癌药单独作用对照组。细胞存活率(%)=A实验组/ADMSO组×100%。MTS检测及存活率计算同细胞毒性检测。
1.4 流式细胞术检测米哚妥林对 P-gp的外排功能
收集K562/A02和K562细胞并计数,细胞按5×108L-1密度以10%FBS的1640培养基悬浮,分装至1.5 ml EP管中待用。按终浓度米哚妥林和维拉帕米加入0.5 和10 μmol·L-1,以0.1%DMSO为阴性对照,37℃恒温水浴30 min。除裸细胞外,各管加入 Rh-123 至终浓度 0.5 μmol·L-1,37℃避光孵育30 min。取出EP管,5000×g离心1 min,去上清,预冷生理盐水洗涤2次,每次1 ml。300 μl预冷的生理盐水重悬细胞,上流式细胞仪检测(通道为FL1-H,激发波长488 nm,发射波长520 nm),逆转倍数=荧光值实验组/荧光值对照组。
1.5 Western印迹法检测米哚妥林对耐药细胞中P-gp的表达
取对数生长期细胞,制成单细胞悬液,按每孔5×105个细胞接种6孔板。接种24 h后,加入化合物继续培养72 h。培养结束,提取细胞总蛋白制备蛋白样品;每孔加入等量待测蛋白,进行SDSPAGE检测(12%);电泳结束,硝酸纤维素膜覆盖,冰浴下60 V转印3 h;5%脱脂奶室温封闭1 h;一抗(1∶1000稀释)4℃孵育过夜;1×TBST洗涤3次,每次10 min;二抗(HRP-GAR,HRP-GAM,1∶2000稀释)室温孵育2 h;1×TBST洗涤3次,每次10 min;加入ECL显色,暗室曝光。
1.6 定量PCR检测米哚妥林对耐药细胞中P-gp基因表达水平
取对数生长期K562/A02细胞,以按每孔5×105细胞接种6孔板,加入米哚妥林至终浓度0.5 μmol·L-1培养 72 h,1‰DMSO 作为阴性对照。培养结束,按照 Trizol试剂说明书提取细胞总RNA,稀释后检测 A260nm和 A280nm,并换算为计算RNA含量。按逆转录试剂盒提供体系进行逆转录,每 20 μl体系含 1 μg RNA。
定量PCR以前述cDNA为模板用SYBR Green Realtime PCR Super Mix按照以下体系进行:SYBR Mix 10 μl,上下游引物各1 μl,cDNA 1 μl,ddH2O 7 μl,总体积共 20 μl。
反应条件如下:① 95℃ 3 min;② 95℃持续30 s,60℃持续20 s 72℃持续20 s并收集荧光共30个循环;③ 从60℃开始以0.5℃为一个梯度逐步升温检测直至95℃,绘制溶解曲线。
MDR1正向引物序列为:5'-AAATTGGCTTGACAAGTTGTATATGG-3'(26 bp),反向:5'-CACCAGCATCATGAGAGGAAGTC-3'(23 bp);GAPDH正向:5'-CCGTCTAGAAAAACCTGCC-3'(19 bp),反向:5'-GCCAAATTCGTTGTCATACC-3'(20 bp)。所有引物由北京诺赛公司合成。
1.7 P-gp-GloTM检测系统检测米哚妥林对P-gp的ATP水解酶活性
P-gp的转运依赖于ATP的水解驱动,化合物与P-gp相互作用会影响ATP酶活性。通过超速离心分离P-gp膜蛋白,并应用P-gp-GloTM检测系统提供的ATP依赖的萤火虫荧光素酶发光效应进行检测。P-gp首先与化合物共孵育,再添加入一定量的ATP,随后终止P-gp ATP酶的反应,剩余未被水解酶代谢的ATP用荧光素酶反应体系被检测。主要步骤如下:① 用试剂盒缓冲液分别稀释米哚妥林、Na3VO4、维拉帕米和DMSO,DMSO终浓度均调整至 1%,使其终浓度为0.2 mmol·L-1,同时将提取的P-gp膜蛋白浓度稀释至 1.5 g·L-1,均放于冰上待用;② 取0.5 ml EP管,标号加入相应化合物,置于冰上,各组有3个复孔。DMSO组加入20 μl 1%DMSO的检测缓冲液;Na3VO4、维拉帕米和米哚妥林各组加入相应20 μl化合物0.1 mmol·L-1(在50 μl体系中,最终工作浓度为 40 μmol·L-1);③ 每管加入 20 μl P-gp 1.5 g·L-1,37℃ 孵育5 min;④EP管取出,置于冰上,每管加10 μl MgATP 25 mmol·L-1,37℃孵育40 min;⑤ 取出EP 管,按标记号加入不透光96孔白板孔内,每孔另加50 μl ATP检测试剂,室温反应20 min后用荧光光度计测量荧光强度。
1.8 统计学分析
2 结果
2.1 米哚妥林对细胞存活的影响
如图1所示,米哚妥林在P-gp高表达的耐药细胞K562/A02及其对应的敏感细胞K562细胞中毒性无显著差异,米哚妥林 0.5 μmol·L-1对两种细胞均无明显杀伤作用,存活率均可达到80%以上。
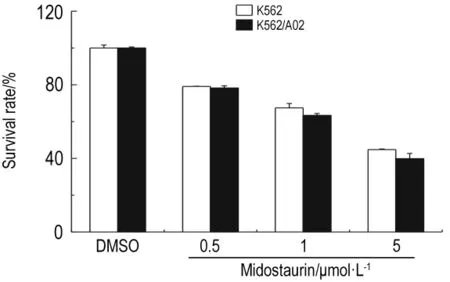
Fig.1 Cytotoxic effect of midostaurin in sensitive and resistant cells by MTS assay.K562/A02 and K562 cells were seeded in 96-well plate followed by addition of midostaurin.Survival rate(%)=AExperiment/AControl×100%.K562/A02 or K562 cells were treated with midostaurin 0.5,1 and 5 μmol·L -1followed by MTS assay.±s,n=3.
2.2 米哚妥林对K562/A02及K562细胞的增敏作用
将无毒剂量的底物类抗癌药与不同浓度梯度的米哚妥林在K562和K562/A02细胞中共同作用评价其逆转MDR的效果,以维拉帕米为阳性对照。结果表明,米哚妥林 0.5 μmol·L-1可显著恢复K562/A02对多柔比星的耐药(图2A),且米哚妥林0.5 μmol·L-1作用效果(细胞存活率 40%)与维拉帕米 2.5 μmol·L-1作用效果接近。对长春新碱(图2B)和紫杉醇(图2C)亦有部分增敏,但效果不如维拉帕米。对亲本敏感细胞K562无效果,表明米哚妥林通过对P-gp的作用起到逆转效果。

Fig.2 Effect of midostaurin on sensitization of drug resistance mediated by P-gp.K562/A02 or K562 cells were treated with doxorubicin(A),vincristine(B),paclitaxel(C),and midostaurin 0,0.25 and 0.5 μmol·L -1added into cells for 24 h.Verapamil was used as positive control drug.±s,n=3.**P<0.01,compared with DMSO control group.
2.3 米哚妥林对P-gp外排功能的影响
如图3所示,米哚妥林可显著增加Rh-123在K562/A02细胞中的积累,表明其对P-gp的外排活性具有抑制作用。同时,米哚妥林0.5 和10 μmol·L-1效果均优于维拉帕米。而在K562细胞中则无抑制作用。
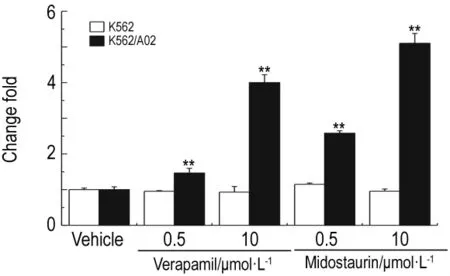
Fig.3 Effect of midostaurin on the intracellular Rh-123 accumulation.Rh-123 accumulation in K562/A02 or K562 cell following a 30 min incubation in the absence or presence of midostaurin or verapamil 0.5 and 10 μmol·L -1 .0.1%DMSO was taken as control and the results were presented as change fold of fluorescence intensity in treatment group relative to DMSO vehicle.±s,n=3.**P<0.01,compared with vehicle control group.
2.4 米哚妥林对P-gp基因与蛋白表达水平的影响
图4 结果表明,米哚妥林0.5 μmol·L-1作用72 h后,在K562/A02细胞中的米哚妥林对P-gp蛋白(图4A)和基因(图4B)均无显著影响。
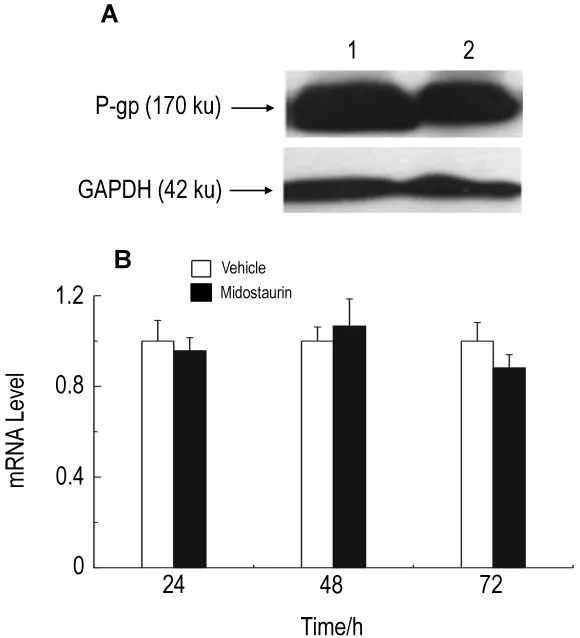
Fig.4 Effect of midostaurin on human P-gp protein expression by Western boltting(A)and mRNA expression by qPCR(B).Lane 1:vehicle control;lane 2:midostaurin 0.5 μmo·lL-1.±s,n=3.
2.5 米哚妥林对耐药和敏感细胞中信号通路分子的影响
米哚妥林的作用靶点包括PKC,FLT3和KIT,可作用于Raf/MEK/ERK通路。通过检测了逆转条件下米哚妥林K562和K562/A02中AKT和ERK的表达及磷酸化水平变化,以探索其逆转耐药的机制是否与其激酶抑制活性相关。如图5所示,米哚妥林0.5 μmol·L-1处理72 h 后对耐药和敏感细胞中 AKT和ERK的表达及磷酸化水平均无显著影响。
2.6 米哚妥林对P-gp的ATP水解酶活性的影响
与对照组相比,荧光信号的减弱表示被P-gp ATP酶消耗的ATP量,因此信号降低的越多,P-gp ATP酶活性越高,可认为P-gp的底物。反之,信号的增强表明,ATP酶活性被抑制,化合物P-gp的为抑制剂。在本实验中,试剂盒提供的Na3VO4为抑制剂对照,维拉帕米为底物对照。如图6所示,米哚妥林不激活P-gp的ATP酶活性,可能不是ATP酶的底物。

Fig.6 Effect of midostaurin on P-gp ATPase activity.Crude membranes from K562/A02 cells were incubated with DMSO or midostaurin 40 μmol·L -1diluted with buffer solution and MgATP 5 mmo·lL-1at 37℃for 40 min.Relative light units(RLU)represent the level of ATP in the sample,exhibiting a negative relationship with activity of P-gp ATPase.Sodium vanadate(Vi)and topotecan(TOPT)40 μmol·L -1were used as inhibitor and substrate control respectively.±s,n=3.*P<0.05,** P<0.01,compared with vehicle group.
3 讨论
P-gp是于20世纪70年代发现的首个耐药相关蛋白,在结肠癌、直肠癌与肾癌等肿瘤中可原发性高表达,在急性白血病、淋巴瘤、小细胞肺癌、卵巢癌等肿瘤化疗中可获得性表达,临床资料显示P-gp过表达与患者的不良预后有关[11]。P-gp的底物主要包括蒽环类物 (多柔比星和柔红霉素)、植物碱(长春新碱和长春碱)、紫杉烷(紫杉醇和多西紫杉醇)类普遍在临床上使用的肿瘤化疗药物[12],而近年的研究表明,P-gp亦有可能与伊马替尼等多种新型激酶抑制剂的临床耐药相关[5]。
近30年来,一直致力于对多药耐药逆转剂的开发。以维拉帕米、环菌素A等第一代逆转剂为代表,其特征多为P-gp底物,依靠与其他药物竞争结合并外排发挥抑制作用,但由于其与P-gp亲和力较低,一般使用剂量较大,并且伴有明显的毒性作用[13]。通过对第一代逆转剂进行结构改造,发展了以维拉帕米右旋同分异构体、PSC833(环孢素A衍生物)和VX-710为代表的第二代逆转剂。与第一代相比,它们的主要特点是具有较低的细胞毒作用,且逆转MDR作用明显增强,但限制其临床应用的主要障碍是与抗肿瘤药物合用时干扰后者的药代动力学[14]。目前以高效低毒、高亲和力、非P-gp底物、无明显药代动力学改变为主要特征的第三代多药耐药逆转剂正在研发中。
本研究中,米哚妥林 0.5 μmol·L-1(IC20以内)即可在功能水平有效逆抑制P-gp的外排活性,在细胞水平可逆转P-gp介导的MDR,表现出较好的逆转活性,同时在ATP酶实验中表明其可能不是P-gp的底物。而已有报道表明米哚妥林临床使用其血药浓度可达到 771~4649 nmol·L-1[15],大于本研究采用的 0.5 μmol·L-1。这表明了这一逆转策略应用的可行性。但是,近期研究表明米哚妥林在肝中需通过CYP3A4酶进行代谢[16]。这使得米哚妥林这种新型的激酶抑制剂是否能作为P-gp抑制剂直接开发应用还有待体内进一步研究,因为米哚妥林具有P-gp抑制作用和CYP3A4底物的特性,这表明其很可能会影响其他药物的药代动力学,在与其他化疗药物(特别是P-gp底物)联用时要在用药的剂量、时序等方面给予特别的重视。但是,鉴于目前投入临床试验的化合物多是在已知的活性化合物骨架结构的基础上改造、修饰通过对其衍生物药效学筛选得到,而至今仍没有一种小分子P-gp抑制剂被投入临床应用,因此,人们需要寻找更多全新结构的化合物,为未来多药耐药逆转剂的开发提供更多空间。所以米哚妥林以其前述特征可作为一种P-gp逆转剂的先导化合物骨架可考虑列入。
[1]Sharom FJ.ABC multidrug transporters:structure,function and role in chemoresistance[J].Pharmacogenomics,2008,9(1):105-127.
[2]Choudhuri S,Klaassen CD.Structure,function,expression,genomic organization,and single nucleotide polymorphisms of human ABCB1(MDR1),ABCC(MRP),and ABCG2(BCRP)efflux transporters[J].Int J Toxicol,2006,25(4):231-259.
[3]Baselga J.Targeting tyrosine kinases in cancer:the second wave[J].Science,2006,312(5777):1175-1178.
[4]Hegedus C,Ozvegy-Laczka C,Szakács G,Sarkadi B.Interaction of ABC multidrug transporters with anticancer protein kinase inhibitors:substrates and/or inhibitors[J]?Curr Cancer Drug Targets,2009,9(3):252-272.
[5]Burger H,van Tol H,Brok M,Wiemer EA,de Bruijn EA,Guetens G,et al.Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2(BCRP)and ABCB1(MDR1)drug transport pumps[J].Cancer Biol Ther,2005,4(7):747-752.
[6]Dohse M,Scharenberg C,Shukla S,Robey RW,Volkmann T,Deeken JF,et al.Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib,nilotinib,and dasatinib[J].Drug Metab Dispos,2010,38(8):1371-1380.
[7]Agarwal S,Sane R,Gallardo JL,Ohlfest JR,Elmquist WF.Distribution of gefitinib to the brain is limited by P-glycoprotein(ABCB1)and breast cancer resistance protein(ABCG2)-mediated active efflux[J].J Pharmacol Exp Ther,2010,334(1):147-155.
[8]Dai CL,Tiwari AK,Wu CP,Su XD,Wang SR,Liu DG,et al.Lapatinib(Tykerb,GW572016)reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2[J].Cancer Res,2008,68(19):7905-7914.
[9]Mi YJ, Liang YJ, Huang HB, Zhao HY,Wu CP,Wang F,et al.Apatinib(YN968D1)reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters[J].Cancer Res,2010,70(20):7981-7991.
[10]Stone RM, Fischer T, Paquette R,Schiller G,Schiffer CA,Ehninger G,et al.Phase ⅠB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia[J].Leukemia,2012,26:2061-2068.
[11]Leighton JC Jr, Goldstein LJ. P-glycoprotein in adult solid tumors.Expression and prognostic significance[J].Hematol Oncol Clin North Am,1995,9(2):251-273.
[12]Flens MJ,Zaman GJ,van der Valk P,Izquierdo MA,Schroeijers AB,Scheffer GL,et al.Tissue distribution of the multidrug resistance protein[J].Am J Pathol,1996,148(4):1237-1247.
[13]Szakács G,Paterson JK,Ludwig JA,Booth-Genthe C,Gottesman MM.Targeting multidrug resistance in cancer[J].Nat Rev Drug Discov,2006,5(3):219-234.
[14]Zhao Q,Li Y,Peng H.Structure basis of P-gp-ligands interaction and reversal of P-gp mediated multidrug resistance[J],J Int Pharm Res(国际药学研究杂志),2010,37:439-445.
[15]Millward MJ,House C,Bowtell D,Webster L,Olver IN,Gore M, et al. The multikinase inhibitor midostaurin(PKC412A)lacks activity in metastatic melanoma:a phaseⅡA clinical and biologic study[J].Br J Cancer,2006,95(7):829-834.
[16]Wang Y, Yin OQ, Graf P, Kisicki JC,Schran H.Dose-and time-dependent pharmacokinetics of midostaurin in patients with diabetes mellitus[J].J Clin Pharmacol,2008,48(6):763-775.
猜你喜欢
云南化工(2021年6期)2021-12-21 07:30:56
科学(2020年2期)2020-08-24 07:57:00
生物技术通报(2015年1期)2015-04-10 16:15:19
南方周末(2014-09-25)2014-09-25 01:43:01
中国药业(2014年21期)2014-05-26 08:56:28
中国药业(2014年19期)2014-05-17 03:11:57
计算物理(2014年1期)2014-03-11 17:00:14
中外医疗(2013年16期)2013-02-01 21:49:07
天津医科大学学报(2011年4期)2011-07-13 09:56:06
中国合理用药探索(2011年3期)2011-03-20 16:30:16
