
蒽和菲加氢反应热力学分析
2013-11-05 05:35侯朝鹏李永丹夏国富李明丰
石油化工 2013年7期
侯朝鹏,李永丹,夏国富,李明丰
(1. 中国石化 石油化工科学研究院,北京 100083;2. 天津大学 化工学院,天津 300072)
近年来,炼油厂加工高硫、重质原油的比例不断增大,如何高效利用石油资源并实现绿色可持续发展,成为人们日益关注的问题。由于重质原油含有大量的多环芳烃,且随环保法规的日趋严格,加氢工艺作为一种重要的重质原油轻质化技术越来越受到人们的重视。
在较缓和的加氢条件下,多环芳烃加氢比相应的单环芳烃加氢更容易。多于一个苯环的芳烃化合物的加氢反应是通过逐步反应进行的,其中每一步反应都可逆,且在多环芳烃的加氢过程中,一般情况下,第一个芳烃加氢的平衡常数(K)最大[1]。过去直接用于计算芳烃加氢平衡常数的实验数据很少[2-3],研究者常根据基团贡献法估算反应的平衡常数,但该方法的计算误差较大。Girgis等[4]曾指出,由于估算方法的不准确性,计算的平衡常数的误差可能大于一个数量级。Frye等[5-6]采用气相体系测定了不同温度、压力下,联苯、茚、萘、菲、二氢苊和芴等几种芳烃与氢气混合物的平衡组成。实验结果表明,当加氢反应温度高于340 ℃时,常压下加氢反应平衡常数小于1。为使芳烃加氢反应具有工业意义,该反应必须在氢压较高的条件下操作。他们的计算结果表明,当氢压由9.7 MPa增至13.7 MPa时,396 ℃下萘的平衡转化率从17%增至84%。在传统的加氢条件下,多环芳烃第一个芳环的加氢平衡常数一般较高[1,7-8]。但以上文献均没有考虑实际的反应过程和复杂的实验条件。在研究过程中,通常选取蒽和菲及其同系物为三环和多环芳烃的模型反应物[9-22]。
针对以上问题,本工作利用热力学数据库软件,对蒽和菲加氢反应热力学平衡进行了讨论,考察了反应温度和体系压力对芳烃加氢转化率的影响,为多环芳烃加氢反应过程提供相关操作条件。
1 芳烃加氢饱和热力学计算依据及数据基础
利用商业软件HSC-Chemistry4.0进行热力学计算,各物质的热力学数据由商用软件的数据库引出。所有气体均认为是理想气体,所有反应以每摩尔主反应物为基准,并假定所有反应体系均为封闭体系。压力指绝对压力,计算结果均为达到化学平衡时的组成,转化率为平衡转化率。蒽和菲的加氢产物较复杂,为表达方便,本实验采用含氢基的平衡组成对这些过程进行描述,这样处理也较接近实际情况。
2 计算结果
2.1 蒽加氢反应体系
多环芳烃加氢的模型反应物通常为蒽和菲[10-16]。气相蒽的加氢产物有多种,为简化描述,主要考虑部分加氢产物四氢蒽和饱和加氢产物全氢蒽两种,其中全氢蒽有cis-trans-式和transsyn-trans-式两种同分异构体,分别简写为ct-全氢蒽和tst-全氢蒽。在标准状况下,蒽加氢生成四氢蒽和全氢蒽的化学反应方程式见式(1)~(5)。

由方程式(1)~(5)可看出,蒽加氢反应为强放热反应,降低温度在热力学上对蒽加氢有利。另外蒽加氢反应为体积缩小反应,增加压力有利于提高蒽转化率,同时提高氢气分压也有利于蒽的转化。
在一个标准大气压下,蒽加氢体系的反应自由能(ΔG)和lgK随温度的变化情况见图1。由图1a可见,蒽加氢反应体系的共同特点是:在100~400℃内,ΔG随温度的升高而近似线性的增加;当温度低于250 ℃时,ΔG为负值,表明这些反应在低于250 ℃时为自发过程,且温度越低,ΔG越小。由图1a还可见,随温度的变化,这5个反应的ΔG的变化趋势有差别,其中蒽加氢生成四氢蒽(反应方程式(1))的ΔG随温度的变化率最小。
由图1b可见,随温度的升高,lgK单调降低。在5个反应中,当温度高于250 ℃时,蒽加氢生成四氢蒽的lgK最大,但当温度低于250 ℃时,蒽加氢生成四氢蒽的lgK又最小。
因为大多数的加氢反应是在高氢烃比的条件下进行的,因此在氢与烃摩尔比为10时,计算了蒽加氢体系的平衡组成与温度和压力的关系,计算结果见图2。由图2a可见,低温区域,在蒽加氢的平衡体系中,蒽含量为零,表明低温在热力学上对蒽加氢反应有利。

图2 温度和压力对蒽加氢产物分布的影响Fig.2 Effects of temperature and pressure on the equilibrium composition of the products in the anthracene hydrogenation system.n(H2)∶n(Aromatic)=10.
在蒽加氢体系中,对应于不同压力,随温度的升高,体系中蒽的平衡含量不断增加,即蒽的平衡转化率随温度的升高而降低。但随压力的升高,蒽平衡含量较低的温度区域逐渐变宽,说明升高压力有利于蒽加氢反应的进行。如在0.1 MPa下,蒽接近完全转化的温度约为200 ℃;而在5.0 MPa时,蒽接近完全转化的温度升至350 ℃。
由图2b可见,在该体系中,对应于不同压力,在较低温度时,随温度的升高,四氢蒽的平衡含量均逐渐增加;当温度超过某一值时,四氢蒽的平衡含量又逐渐减小,在此期间存在一个最大值;当压力为0.1 MPa时,该最大值在250 ℃附近。随压力的升高,四氢蒽的最大平衡含量逐渐向高温偏移。由图2c可见,对应于不同压力,随温度的升高,tst-全氢蒽的平衡含量均逐渐减少。但随压力的升高,生成tst-全氢蒽的温度区域逐渐变宽,说明升高压力有利于tst-全氢蒽的生成,高压下蒽加氢的可操作温度更宽。由图2d可见,在该体系中,对应于不同压力,随温度的升高,ct-全氢蒽的平衡含量存在一个最大值;当压力为0.1 MPa时,ct-全氢蒽的最大平衡含量出现在200 ℃左右。随压力的升高,ct-全氢蒽的最大平衡含量逐渐向高温偏移,且在平衡组成中所占的比例逐渐增加。通过比较图2c和2d可见,在热力学上,蒽加氢生成tst-全氢蒽比生成ct-全氢蒽更有利,如在0.1 MPa下,体系中tst-全氢蒽的平衡含量最高可达23.8%(x),而ct-全氢蒽的平衡含量最高只有2.8%(x)。因此,在以气相蒽为模型反应物的芳烃加氢体系中,必须考虑温度、压力和氢烃比等因素,以得到合适的操作条件。
2.2 菲加氢反应体系
菲作为中间馏分油的组成成分,在中间馏分油加氢的研究中,常用作模型反应物[17-22]。气相菲的加氢产物一般有部分加氢的四氢菲和全部加氢的全氢菲两种,其中全氢菲有cis-anti-trans-式、cis-syn-trans-式、trans-anti-trans-式和trans-syncis-式4种同分异构体,分别简写为cat-全氢菲、cst-全氢菲、tat-全氢菲和tsc-全氢菲。
由于全氢菲的同分异构体较多,因此菲加氢体系中的产物分布与蒽加氢的有所不同。本实验仅选择四氢菲和cat-全氢菲进行研究,反应方程式见(6)~(8)。

在一个标准大气压下,菲加氢体系的ΔG和lgK随温度的变化情况见图3。由图3a和b可见,菲加氢反应的ΔG和lgK随温度的变化情况与蒽加氢相似。由图3a可见,在100~400 ℃内,ΔG随温度的升高而近似线性的增加;当温度低于250 ℃时,ΔG为负值,表明这3个反应在低于250 ℃时为自发过程,且温度越低,ΔG越小,反应越倾向于自发进行。由图3a还可见,这3个反应的ΔG随温度变化的趋势有差别,其中菲加氢生成四氢菲的ΔG随温度的变化率最小。
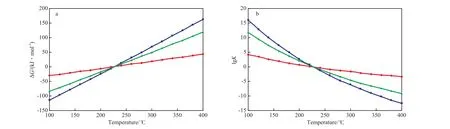
图3 菲加氢体系的ΔG(a)和lgK(b)随温度的变化曲线Fig.3 ΔG(a) and lgK(b) vs temperature in phenanthrene hydrogenation.
由图3b可见,随温度的升高,lgK单调降低;在这3个反应中,当温度高于250 ℃时,菲加氢生成四氢菲的lgK最大;但当温度低于250 ℃时,菲加氢生成四氢菲的lgK又最小。
由于全氢菲的同分异构体较多,在三维图中画出其平衡组成的变化情况较复杂,因此选择了特定条件进行比较,实验结果见图4。
由图4a可见,在0.1 MPa下,当温度低于200℃时,菲的平衡转化率很高,产物主要为4种同分异构的全氢菲。随温度的升高,菲的平衡转化率降低。部分加氢产物四氢菲的平衡含量随温度的升高先增大后降低,在温度为250 ℃左右时出现最大值。由图4a还可见,tat-全氢菲的平衡含量最高,说明热力学上tat-全氢菲比其他全氢菲更易自发生成;对于全氢菲,当温度低于210 ℃时,随温度的升高,tat-全氢菲的平衡含量有所降低,而其他全氢菲的平衡含量则有所提高;当温度高于210 ℃后,所有全氢菲的平衡含量都降低。
由图4b可见,在压力为5.0 MPa时,菲加氢体系中的平衡组成随温度的变化趋势与压力为0.1 MPa时的类似,但整体范围向高温方向移动。当温度低于350 ℃时,菲的平衡转化率很高,与0.1 MPa时相比,温度范围加宽约150 ℃,表明压力对平衡组成的影响非常大。随温度的升高,菲的平衡转化率降低。部分加氢产物四氢菲的平衡含量随温度的升高先增大后减小,在温度为400 ℃左右时有最大值。另外对于全氢菲,当温度低于350 ℃时,随温度的升高,tat-全氢菲的平衡含量有所降低,而其他全氢菲的平衡含量则有所提高。当温度高于350℃后,全氢菲4种异构体的平衡含量均降低。
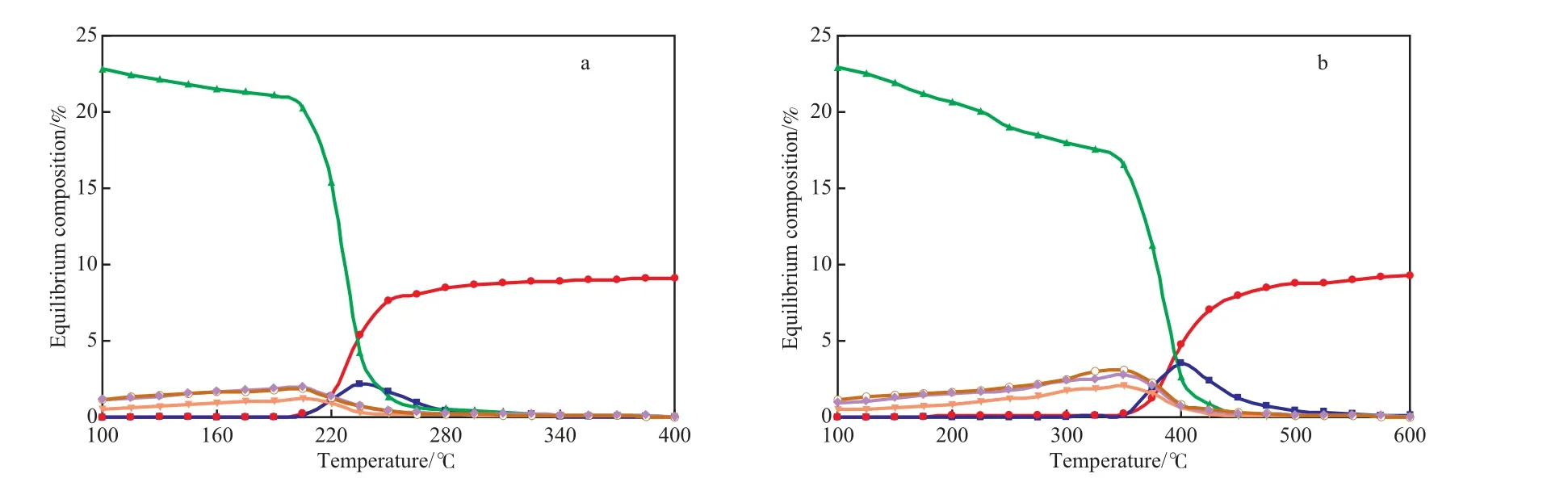
图4 温度对菲加氢产物分布的影响Fig.4 Effects of temperature on the equilibrium composition of the products in the phenanthrene hydrogenation.n(H2)∶n(Aromatic)=10.
3 结果与讨论
3.1 计算结果的参考意义
虽然本实验没有对各种芳烃及其所有加氢产物的热力学计算进行一一考虑,但所选芳烃和加氢产物都是生产和实验研究中经常使用的典型模型反应物[10-26]。以上计算结果有助于研究者寻找典型芳烃加氢在热力学上合适的操作区域,作为实验中选择反应条件的依据。对于实际操作过程,可选择它们的合适操作区域的公共部分,在热力学上保证不同芳烃加氢过程的可行性。特别是在实验操作过程中,一般会针对反应过程选择具体的模型反应物,而这些典型芳烃的热力学计算会直接提供相关的热力学上所允许的反应温度、氢烃比和体系压力等操作条件,从而简化整个研究过程。
3.2 操作条件对芳烃加氢的影响
温度是影响芳烃加氢反应热力学平衡的重要因素。从前面的热力学计算可知,芳烃加氢反应是可逆反应,由于反应是强放热过程,因此芳烃加氢平衡转化率随温度的升高而降低。虽然低温从热力学平衡角度上有利于芳烃的加氢转化,但从动力学角度上讲,温度不能太低,否则反应速率太慢,因此,必须针对工艺选取合适的操作温度。
在常规的加氢条件下,多环芳烃加氢反应速率很慢,一般需在300 ℃以上的高温下进行,但高温时热力学对芳烃加氢的限制起主导作用,反应平衡常数变小,所以,在使用硫化态的金属催化剂时,馏分油中的芳烃(特别是单环芳烃)加氢饱和较困难,加氢深度也较低。而采用高活性的金属催化剂,芳烃加氢反应的活化能较低,加氢反应可在低温下进行,在热力学上几乎不受限制,反应平衡常数也很大,有利于提高芳烃的转化率[25-26]。
在实际操作和工业生产中,要充分考虑工作中的问题。如在反应过程中出现热点会使反应床层的温度升高,不利于芳烃的转化,但此时在反应过程中提高压力和氢烃比,则可使反应的高芳烃转化率的可操作温度范围变宽,从而保证生产的顺利进行。又如,在生产过程中,催化剂会出现部分失活,有时需提高反应温度使生产进行下去;要生产低芳烃含量的油品和溶剂时,则必须适当调整反应体系的压力和氢烃比[1,25-26]。
4 结论
1)选取蒽和菲为模型反应物,对蒽和菲加氢反应热力学的可能性和反应体系的平衡组成进行了讨论,考察了反应条件对蒽和菲加氢转化率的影响。低温和高压在热力学上对蒽和菲加氢有利,蒽和菲的平衡转化率随温度的升高而降低。
2)在蒽加氢体系中,四氢蒽的平衡含量存在一个最大值,且随压力的升高,四氢蒽的最大平衡含量向高温偏移。随温度的升高,tst-全氢蒽的平衡含量降低,ct-全氢蒽的平衡含量也存在一个最大值。在热力学上蒽加氢生成tst-全氢蒽比生成ct-全氢蒽更有利。
3)在菲加氢体系中,随温度的升高,菲的平衡转化率降低,四氢菲的平衡含量存在一个最大值。热力学上生成tat-全氢菲比生成其他全氢菲明显有利,tat-全氢菲的平衡含量最高。压力对平衡组成和有效温度操作范围的影响非常大。
4)这些计算不但直接提供了蒽和菲加氢过程的热力学可行性,而且为选择多环芳烃加氢过程的操作参数和条件提供了参考和指导。
[1] 李大东. 加氢处理工艺与工程[M]. 北京:中国石化出版社,2004:119 - 129.
[2] Shaw R,Golden D M,Benson S W. Thermochemistry of Some Six-Membered Cyclic and Polycyclic Compounds Debated to Coal[J]. J Phys Chem,1977,81(18):1716 - 1729.
[3] Stein S,Golden D M,Benson S W. Prediction Scheme for Thermodynamical Properties of Polycyclic Aromatic Hydrogenation[J]. J Phys Chem,1977,81(4):314 - 316.
[4] Girgis M J,Gates B C. Reactivities,Reaction Networks,and Kinetics in High-Pressure Catalytic Hydroprocessing[J]. Ind Eng Chem Res,1991,30(9):2021 - 2058.
[5] Frye C G. Equilibria in the Hydrogenation of Polycyclic Aromatics[J]. J Chem Eng Data,1962,7(4):592 - 595.
[6] Frye C G,Weitkamp A W. Equilibrium Hydrogenations of Multi-Ring Aromatics[J]. J Chem Eng Data,1969,14(3):372 - 376.
[7] Le Page J F. Catalyse de Contact:Conception,Preparation et mise en æuvre des Catalyseurs Industriels[M]//李宣文,黄志渊,译.接触催化:工业催化剂原理、制备及其应用. 北京:石油工业出版社,1984:7.
[8] Stanislaus A,Copper B H. Aromatic Hydrogenation Catalysis:A Review,Catalysis Review[J]. Sci Eng,1994,36(1):75 - 123.
[9] 何国锋,关北峰,王燕芳,等. 温热解焦油中油馏分加氢脱氮和芳烃加氢宏观动力学的研究[J]. 煤炭转化,1998,21(1):54 - 58.
[10] 张全信,刘希尧. 多环芳烃的加氢裂化[J]. 工业催化,2001,9(2):10 - 16.
[11] Zhang Z G,Okada K,Yamamoto M,et al. Hydrogenation of Anthracene over Active Carbon-Supported Nickel Catalyst[J]. Catal Today,1998,45(1/4):361 - 366.
[12] Kotanigawa T,Yamamoto M,Yoshida T. Selective Nuclear Hydrogenation of Naphthalene,Anthracene and Coal-Derived Oil over Ru Supported on Mixed Oxide[J]. Appl Catal,A,1997,164(1/2):323 - 332.
[13] 刘会茹,徐智策,赵地顺. 多环芳烃在贵金属催化剂上竞争加氢反应的研究[J]. 化学学报,2007,65(18):1933 -1939.
[14] Alvarez J,Rosal R,Sastre H,et al. Characterization and Deactivation Studies of an Activated Sulfided Red Mud Used as Hydrogenation Catalyst[J]. Appl Catal,A,1998,167(2):215 - 223.
[15] Sidhpuria K B,Patel H A,Parikh P A,et al. Rhodium Nanoparticles Intercalated into Montmorillonite for Hydrogenation of Aromatic Compounds in the Presence of Thiophene[J].Appl Clay Sci,2009,42(3/4):386 - 390.
[16] Jacinto M J,Santos O H C F,Landers R,et al. On the Catalytic Hydrogenation of Polycyclic Aromatic Hydrocarbons into Less Toxic Compounds by a Facile Recoverable Catalyst[J].Appl Catal,B,2009,90(3/4):688 - 692.
[17] Nuzzi M,Marcandalli B. Hydrogenation of Phenanthrene in the Presence of Ni Catalyst:Thermal Dehydrogenation of Hydrophenanthrenes and Role of Individual Species in Hydrogen Transfers for Coal Liquefaction[J]. Fuel Process Technol,2003,80(1):35 - 45.
[18] Qian Weihua,Yoda Yosuke,Hirai Yoshiki,et al. Hydrodesulfurization of Dibenzothiophene and Hydrogenation of Phenanthrene on Alumina-Supported Pt and Pd Catalysts[J]. Appl Catal,A,1999,184(1):81 - 88.
[19] Menini R,Martel A,Me´nard H,et al. The Electrocatalytic Hydrogenation of Phenanthrene at Raney Nickel Electrodes:The Inf l uence of an Inert Gas Pressure[J]. Electrochim Acta,1998,43(12/13):1697 - 1703.
[20] 张聪琳,吴倩,李佟茗. 菲加氢裂化催化剂的初步研究[J].化工时刊,2009,23(8):4 - 7.
[21] 李会峰,刘锋,刘泽龙,等. 菲在不同加氢催化剂上的转化[J]. 石油学报:石油加工,2011,27(1):20 - 25.
[22] Leite L,Benazzi E,Marchal-George N. Hydrocracking of Phenanthrene over Bifunctional Pt Catalysts[J]. Catal Today,2001,65(2/4):241 - 247.
[23] 侯朝鹏. FC制备镍金属催化剂苯加氢反应和抗噻吩能力的研究[D]. 天津:天津大学,2003.
[24] 路长通,谷双,陈纪忠. 热重模拟蒸馏在重油馏程分析中的应用[J]. 石油化工,2011,40(9):1006 - 1009.
[25] 侯镜德,路文初,周瑛. 基团贡献法关联多环芳烃的反向高效液相色谱保留值[J]. 石油化工,1997,26(7):457 - 459.
[26] 侯朝鹏,李永丹,赵地顺. 芳烃加氢金属催化剂抗硫性研究的进展[J]. 化工进展,2003,22(4):366 - 371.
猜你喜欢
中学生数理化(高中版.高考理化)(2021年10期)2021-12-06
科学家(2021年24期)2021-04-25
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
上海金属(2016年1期)2016-11-23
电子制作(2016年19期)2016-08-24
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
山西大同大学学报(自然科学版)(2015年1期)2015-01-22
汽车与新动力(2014年4期)2014-02-27
