
类微乳化学沉淀法合成BiOBr-TiO2纳米复合材料及其光催化性能
2013-10-18 05:27栾云博冯玉杰王文欣谢明政井立强
物理化学学报 2013年12期
栾云博 冯玉杰,* 王文欣 谢明政 井立强,*
(1哈尔滨工业大学,城市水资源与水环境国家重点实验室,城市水资源开放利用(北方)工程研究中心,哈尔滨 150090;2黑龙江大学,功能无机材料化学教育部重点实验室,哈尔滨 150080)
1 引言
半导体光催化氧化技术因具有操作简单、反应条件温和、能耗低及二次污染少等优点,在环境治理领域备受关注.1,2在众多的半导体光催化剂中,TiO2因具有高化学活性、强氧化能力,无毒及在光和化学物质的侵蚀下的高稳定性的优点,成为理想的光催化剂材料之一.3-5但是,TiO2是宽禁带半导体(锐钛矿TiO2的Eg=3.2 eV),对太阳光的利用率低;同时,光激发产生的电子和空穴的复合率高,光量子效率差.6,7这两个瓶颈因素限制了它的实际应用.因此,对TiO2进行改性研究提高其光催化活性成为当前研究的热点.在TiO2的改性研究中,窄带隙半导体材料与宽禁带半导体TiO2进行复合是重要的研究课题.8,9BiOBr作为一种新型的窄带隙半导体材料拥有独特的电子结构、良好的光学性能和催化性能等优点,因此吸引了研究者的广泛兴趣.10-12然而,目前文献将BiOBr与TiO2复合获得高性能光催化剂材料的相关研究报道较少.
利用两种半导体材料复合方法提高TiO2光量子效率的关键在于两种半导体材料之间有效异质结的构筑.通常人们在水相中采用浸渍法、共沉淀法或溶胶-凝胶法等实现两种半导体材料的复合.13,14而这些方法不仅难以控制窄带隙半导体的粒子尺寸和形貌,同时也会影响两种半导体之间异质结的有效构筑,进而影响着复合半导体光催化材料的电荷传输以及光催化性能.因此,当今亟需一种新的合成方法来解决上述问题.目前,人们常常选用十六烷基三甲基溴化铵(CTAB)作为溴源,在高温溶剂热条件下获得BiOBr光催化剂材料.15-17而CTAB作为一种阳离子表面活性剂又往往被人们用于微乳体系的构筑.18,19如果利用CTAB构筑的TiO2微乳体系的同时,又以该表面活性剂作为溴源制备BiOBr粒子,有望在BiOBr和TiO2之间构筑有效的异质结.此外,我们前期工作20利用该策略成功获得了AgBr-TiO2复合材料,其在降解气相乙醛和液相苯酚无色污染物时表现出高于纯TiO2的光催化活性.因此,我们推测采用此方法将会合成出具有高性能的BiOBr-TiO2光催化剂.然而据我们了解,至今没有采用上述合成策略获得高性能BiOBr-TiO2复合光催化剂的相关报道.
在本文中,我们利用类微乳化学沉淀法获得了能够高效降解气相乙醛和液相苯酚BiOBr-TiO2纳米光催化剂.此合成方法的关键在于CTAB中Br-的双重作用,它既作为“桥”使CTA+修饰在纳米TiO2表面构建了稳定的油包水的类微乳体系,又作为溴源为实现BiOBr与TiO2的有效复合提供条件.并且借用透射电子显微镜和表面光电压技术揭示出类微乳化学沉淀法获得BiOBr-TiO2表现出高于纯TiO2和传统水相化学沉淀法获得的复合材料的光催化性能的原因,主要与该方法能够有效构筑BiOBr与TiO2粒子之间的异质结结构,进而有利于提高其光生载流子的分离效率有关.此工作的开展必将为设计合成高性能的复合纳米光催化剂材料提供思路.
2 实验部分
实验所涉及的药品和试剂均为分析纯且未经过进一步纯化,所用的水均为二次蒸馏水.
2.1 材料合成
2.1.1 TiO2类微乳体系的构筑
首先采用溶胶-水热法合成了二氧化钛纳米粒子.21,22将5 mL钛酸四丁酯和5 mL无水乙醇的均匀混合液缓慢地滴加到20 mL无水乙醇、5 mL水和1 mL浓硝酸的混合液中,剧烈搅拌1 h后,可得到淡黄色的溶胶.将溶胶转移至不锈钢高压反应釜中,在160°C条件下水热6 h,自然冷却至室温,去除上清液即得到淡黄色膏状TiO2.然后,利用1 mol·L-1的硝酸调制出釜的TiO2膏体溶液至pH=2后,再在混合溶液中加入0.0629 g CTAB.经过多次搅拌,超声后,将膏体溶液转移到甲苯中,构建类微乳体系.此外,在对比实验中,采用水代替甲苯溶液,构建水溶液体系.
2.1.2 BiOBr-TiO2复合材料的制备
向已经构筑的TiO2类微乳体系中缓慢滴加0.0837 g Bi(NO3)3·5H2O和5 mL乙二醇(EG)的混合溶液.经过搅拌后,利用水和无水乙醇反复洗涤、光照、离心、干燥后,获得BiOBr-TiO2复合样品.将在类微乳体系下获得的复合样品标记为BT-M,而在水溶液体系下获得的复合样品标记为BT-W.此外,BT-M和BT-W复合样品中BiOBr与TiO2的质量比均为0.05:1.
2.2 材料表征
采用日本理学公司的D/max-IIIB型X射线衍射仪(XRD)测试样品的晶相组成,测试条件为Cu Kα(λ=0.15418 nm),管电压40 kV、管电流30 mA;利用日本JEOL JEM-2010型透射电子显微镜(TEM)观测样品的形貌、粒子尺寸和晶格条纹,电子束加速电压为200 kV;采用日本岛津UV-2550紫外光谱仪测试样品的漫反射(DRS)光学性能;利用自己搭建的表面光电压光谱仪(SPS)对样品的表面光伏性质进行测试.23,24
2.3 光催化活性评估
利用150 W氙灯作为光源,选用气相乙醛和液相苯酚作为目标降解物评估样品的光催化性能.在降解乙醛活性时,将0.1 g催化剂放入由乙醛、氧气和氮气所组成的8.10×10-4L混合气体中,光催化反应1 h后,采用GC-2014型气相色谱仪(氢火焰离子化检测器(FID)检测器)检测乙醛浓度.降解苯酚时,在100 mL的5 mg·L-1苯酚溶液中加入0.4 g光催化剂,光催化反应4 h后,取上层清液离心.用UV-2550型紫外-可见光谱仪以4-氨基安替比林比色法在510 nm波长处测定苯酚浓度,从而确定光催化降解率.
3 结果与讨论
3.1 结构表征
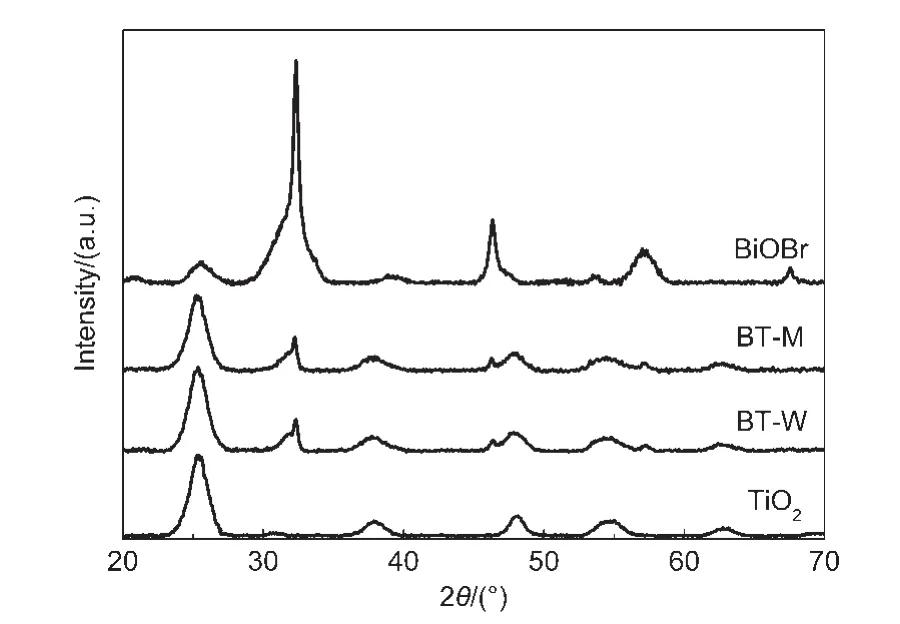
图1 TiO2,BT-W,BT-M和BiOBr样品的XRD图Fig.1 XRD patterns of TiO2,BT-W,BT-M and BiOBr samples
利用XRD测试技术考察了样品的晶相组成和微晶尺寸.由图1可以看出,通过溶胶-水热法获得了锐钛矿相TiO2粒子(JCPDS file:21-1272).利用谢乐公式计算出它的微晶尺寸约为6 nm.25而后采用水相和类微乳化学沉淀法分别制备出的复合样品,都是由四方晶相BiOBr(JCPDS file:01-1004)和锐钛矿相TiO2组成.依据所获得纯样品和复合样品中TiO2和BiOBr的XRD特征峰强度和半峰宽可知,两种方法获得的BiOBr微晶尺寸相近,并且BiOBr的引入不会影响纯TiO2的晶相组成和微晶尺寸.
图2是纳米TiO2和复合样品的TEM照片.从图2a中可以看出,TiO2粒子是由约为6 nm球状颗粒所组成的,这与XRD测试结果相一致.采用水相化学沉淀法获得的BT-W复合样品显示出(如图2b所示),在保持了纯TiO2粒子形貌同时,长约250 nm、宽约50 nm的BiOBr粒子部分与TiO2粒子相连.而从图2c中可以看出,通过类微乳化学沉淀法制备的BT-M复合样品中具有较小粒子尺寸的棒状BiOBr粒子(长约150 nm、宽约40 nm)完全与TiO2纳米粒子相接触.此外,BT-M复合样品的HRTEM照片(图2d)显示出,具有与四方晶相[011]晶面的BiOBr粒子(晶面间距为0.350 nm)和锐钛矿相[101]晶面的TiO2粒子(晶面间距为0.352 nm)紧密接触.26,27这进一步说明,采用类微乳化学沉淀法制备的复合样品中BiOBr和TiO2粒子之间形成了有效的异质结.
3.2 合成机制

图2 TiO2(a),BT-W(b)和BT-M(c)样品的TEM照片以及BT-M样品的高分辨透射电镜(HRTEM)照片(d)Fig.2 TEM images of TiO2(a),BT-W(b)and BT-M(c)samples,and high resolution transmission electron microscopy(HRTEM)image of BT-M(d)
与传统水相化学沉淀法相比,为什么类微乳化学沉淀法构筑的BiOBr与TiO2之间异质结更加有效?在传统水相化学沉淀法中,CTAB与TiO2表面连接趋向于形成胶束.但是,由于CTAB胶束间的空间位阻较大,使得大部分Br-处于游离状态,难以与TiO2表面形成有效的连接.所以,当络合物[BiOCH2CH2OH]2+加入到水体系中,大部分极性端会与游离的Br-生成BiOBr沉淀散落在溶液中,这将影响BiOBr和TiO2粒子之间的有效复合.而在所构筑的类微乳体系中,却与上述情况不同.一般,纳米TiO2的等电点为6.0-6.5.28,29当pH值低于等电点时,纳米TiO2的表面呈正电性.因此,在酸性溶液中,CTAB的亲水端Br-通过氢键或静电作用与纳米TiO2表面相连,而CTAB疏水端进入到甲苯溶液中,从而形成了稳定的类微乳体系.当在此类微乳体系中加入乙二醇和Bi(NO3)3·5H2O混合物后,在极性微核中且与TiO2表面紧密相连的亲水端Br-会迅速和所生成的络合物[BiOCH2CH2OH]2+相互作用,进而形成BrOBi-TiO2复合材料的前驱体.可见,与水体系相比,类微乳体系更加有利于BiOBr与TiO2粒子之间形成有效的异质结,并且此结果充分体现在TEM和HRTEM照片上.BiOBr-TiO2复合材料有效异质结的构筑将有利于提高其光生电荷的分离效率.
3.3 光生电荷性质
图3是纯TiO2和BiOBr-TiO2复合样品的紫外-可见吸收光谱图.由图可知,TiO2样品在小于390 nm波长范围内表现出明显的吸收,这是由TiO2的带带电子跃迁(O 2p→Ti 3d)所引起的.30而采用水相和类微乳化学沉淀法分别合成出的BT-W和BT-M样品均展现出一定的可见光吸收,这主要由于复合样品中含有窄带隙半导体BiOBr.根据Kubelka-Munk法计算出所获得的锐钛矿相TiO2和采用传统化学沉积法制备出纯BiOBr(如图1所示)带隙能分别约为3.18和2.75 eV.31

图3 TiO2,BT-W,BT-M和BiOBr样品的吸收谱图Fig.3 Absorption spectra of TiO2,BT-W,BT-M,and BiOBr samples

Fig.4 TiO2,BT-W和BT-M样品的SPS谱图Fig.4 Surface photovoltage spectroscopy(SPS)responses of TiO2,BT-W,and BT-M samples
半导体材料的表面光电压源于表面和本体之间(或空间电荷区)的光致电荷载流子在自建电场作用下的有效分离,因此也能够反映样品激发状态光生载流子的分离、复合等信息.23,32图4为纯TiO2和BiOBr-TiO2复合样品的表面光电压光谱图(SPS).由图可知,三个样品均在约345 nm处出现了明显的SPS响应信号,根据DRS图谱可知此信号主要由TiO2带带跃迁(O 2p→Ti 3d的电荷迁移跃迁)所引起的.与纯TiO2样品相比,采用水相和类微乳化学沉淀两种方法所获得的BT-W和BT-M复合样品表现出较高的光伏响应信号,其中BT-M表现出最强的光伏响应.一般人们认为,光伏响应信号越高,光生电荷分离效率越好.21,33因此上述结果表明,利用BiOBr与TiO2复合后能够提高纯TiO2的光生电荷分离效率,并且采用微乳化学沉淀法合成出的BT-M拥有最高的光生电荷分离效率.此结果主要归因于BT-M中BiOBr与TiO2之间形成的有效异质结,从而促进了光生电荷载流子的分离.
从光生电荷转移和分离机制图(如图5)可以看到,在光激发下,TiO2价带上的电子激发到导带上,由于BiOBr和TiO2异质结的形成,有利于TiO2导带上的光生电子转移到BiOBr导带上,进而与其表面上吸附的O2反应生成O2-自由基,而光生空穴保留在其价带上,至使TiO2中光生电子和空穴有效分离.与此同时,在光照射下,BiOBr的光生载流子也会被激发.对于被激发的BiOBr粒子而言,导带上的光生电子会与其表面氧气发生还原反应,而在其价带上的光生空穴一部分会转移到位置更高的TiO2价带之上,导致光生载流子进行了有效分离.可见,BiOBr和TiO2异质结的有效构筑有利于其光生载流子的分离,这将会明显提高其光催化活性.
3.4 光催化活性

图6 不同样品的苯酚(A)和乙醛(B)的光催化降解率Fig.6 Photocatalytic degradation rates of phenol(A)and acetaldehyde(B)on different samples
选择液相苯酚溶液和气相乙醛作为目标降解物来评价样品的紫外-可见光催化活性.与光照下降解率相比,样品在暗光下对目标降解物的吸附量是可以忽略不计的.从图6可以看出,与纯TiO2和纯BiOBr样品相比,复合光催化剂都表现出较好的活性,并且在其中采用类微乳法获得的BT-M复合样品表现出最高的光催化降解苯酚和乙醛的性能.这与上述SPS响应信号规律是相一致的,因此,表现出高的光催化活性,这主要是由于类微乳化学沉淀法有利于BiOBr和TiO2粒子之间异质结的构筑进而增加了其光生载流子的分离效率.
4 结论
采用类微乳化学沉淀法获得了BiOBr-TiO2复合材料.与传统水相化学沉淀法相比,类微乳化学沉淀法更有利于BiOBr与TiO2之间异质结的构筑.分析发现,有效异质结的构筑主要与Br-的双重作用有关.它既作为“桥”使CTA+修饰在纳米TiO2表面构建了稳定的油包水的类微乳体系,又作为溴源为实现BiOBr与TiO2粒子之间的有效复合提供了条件.根据SPS测试结果显示,BiOBr-TiO2复合材料粒子之间有效异质结的构筑显著提高了光生电荷分离效率,从而改善了TiO2光催化降解苯酚和乙醛的活性.此工作的开展,将为构筑高性能的溴化物-氧化物复合纳米光催化材料提供可行性的策略.
(1)Fujishima,A.;Honda,K.Nature 1972,238,37.
(2)Hoffmann,M.R.;Martin,S.T.;Choi,W.Y.;Bahnemann,D.W.Chem.Rev.1995,95,69.doi:10.1021/cr00033a004
(3)Asahi,R.;Morikawa,T.;Ohwaki,T.;Aoki,K.;Taga,Y.Science 2001,293,269.doi:10.1126/science.1061051
(4)Nagaveni,K.;Sivalingam,G.;Hegde,M.S.;Madras,G.Environ.Sci.Technol.2004,38,1600.doi:10.1021/es034696i
(5)Li,H.H.;Chen,R.F.;Ma,Z.;Zhang,S.L.;An,Z.F.;Huang,W.Acta Phys.-Chim.Sin.2011,27,1017.[李欢欢,陈润锋,马 琮,张胜兰,安众福,黄 维.物理化学学报,2011,27,1017.]doi:10.3866/PKU.WHXB20110514
(6)Zhao,W.;Ma,W.H.;Chen,C.C.;Zhao,J.C.;Shuai,Z.G.J.Am.Chem.Soc.2004,126,4782.doi:10.1021/ja0396753
(7)Kwak,E.S.;Lee,W.;Park,N.G.;Kim,J.;Lee,H.Adv.Funct.Mater.2009,19,1093.doi:10.1002/adfm.v19:7
(8)Zang,Y.J.;Farnood,R.Appl.Catal.B:Environ.2008,79,334.doi:10.1016/j.apcatb.2007.10.019
(9)Dai,G.P.;Liu,S.Q.;Peng,R.;Luo,T.X.Acta Phys.-Chim.Sin.2012,28,2169.[戴高鹏,刘素芹,彭 荣,罗天雄,物理化学学报,2012,28,2169.]doi:10.3866/PKU.WHXB201207041
(10)Ai,Z.;Ho,W.;Lee,S.;Zhang,L.Environ.Sci.Technol.2009,43,4143.doi:10.1021/es9004366
(11)Zhang,X.;Ai,Z.;Jia,F.;Zhang,L.J.Phys.Chem.C 2008,112,747.doi:10.1021/jp077471t
(12)Yu,C.L.;Cao,F.F.;Shu,Q.;Bao,Y.L.;Xie,Z.P.;Yu,J.C.;Yang,K.Acta Phys.-Chim.Sin.2012,28,647.[余长林,操芳芳,舒 庆,包玉龙,谢志鹏,Yu Jimmy C,杨 凯.物理化学学报,2012,28,647.]doi:10.3866/PKU.WHXB201201051
(13)Ju,J.F.;Li,C.J.;Xu,M.J.Funct.Mater.2005,36,648.[鞠剑峰,李澄俊,徐 铭.功能材料,2005,36,648.]
(14)Li,F.B.;Gu,G.B.;Li,X.J.;Wan,H.F.Acta Phys.-Chim.Sin.2000,11,997.[李芳柏,古国榜,李新军,万洪富.物理化学学报.2000,11,997.]doi:10.3866/PKU.WHXB20001108
(15)Zhang,L.;Cao,X.F.;Chen,X.T.;Xue,Z.L.J.Colloid Interface Sci.2011,354,630.doi:10.1016/j.jcis.2010.11.042
(16)Liu,Z.S.;Wu,B.T.;Zhu,Y.B.;Wang,F.;Wang,L.G.J.Colloid Interface Sci.2013,392,337.doi:10.1016/j.jcis.2012.09.062
(17)Xiao,P.P.;Zhu,L.L.;Zhu,Y.C.;Qian,Y.T.J.Solid State Chem.2011,184,1459.doi:10.1016/j.jssc.2011.04.015
(18)Li,W.W.;Matyjaszewski,K.Polym.Chem.2012,3,1813.doi:10.1039/c1py00431j
(19)Nishioka,K.;Niidome,Y.;Yamada,S.Langmuir 2007,23,10353.doi:10.1021/la7015534
(20)Wang,W.X.;Jing,L.Q.;Qu,Y.C.;Luan,Y.B.;Fu,H.G.;Xiao,Y.C.J.Hazard.Mater.2012,243,169.doi:10.1016/j.jhazmat.2012.10.017
(21)Jing,L.Q.;Qu,Y.C.;Su,H.J.;Yao,C.H.;Fu,H.G.J.Phys.Chem.C 2011,115,12375.doi:10.1021/jp203566v
(22)Luan,Y.B.;Jing,L.Q.;Xie,M.Z.;Shi,X.;Fan,X.X.;Cao,Y.;Feng,Y.J.Phys.Chem.Chem.Phys.2012,14,1352.doi:10.1039/c1cp22907a
(23)Jing,L.Q.;Sun,X.J.;Shang,J.;Cai,W.M.;Xu,Z.L.;Du,Y.G.;Fu,H.G.Sol.Energg Mat.Sol.Cells 2003,79,133.doi:10.1016/S0927-0248(02)00393-8
(24)Lin,Y.H.;Wang,D.J.;Zhao,Q.D.;Yang,M.;Zhang,Q.L.J.Phys.Chem.B 2004,108,3202.doi:10.1021/jp037201k
(25)Zhang,Q.H.;Gao,L.;Guo,J.K.Appl.Catal.B 2000,26,207.doi:10.1016/S0926-3373(00)00122-3
(26)Cheng,H.F.;Huang,B.B.;Wang,Z.Y.;Qin,X.Y.;Zhang,X.Y.;Dai,Y.Chem.Eur.J.2011,17,8039.doi:10.1002/chem.v17.29
(27)Yang,X.H.;Li,Z.;Sun,C.H.;Yang,H.G.;Li,C.Z.Chem.Mater.2011,23,3486.doi:10.1021/cm2008768
(28)Qu,Y.C.;Wang,W.X.;Jing,L.Q.Appl.Surf.Sci.2010,257,151.doi:10.1016/j.apsusc.2010.06.054
(29)Gun ko,V.M.;Zarko,V.I.;Turov,V.V.;Leboda,R.;Chibowski,E.;Pakhlov,E.M.;Goncharuk,E.V.;Marciniak,M.;Voronin,E.F.;Chuiko,A.A.J.Colloid Interface Sci.1999,220,302.doi:10.1006/jcis.1999.6481
(30)Jing,L.Q.;Fu,H.G.;Wang,B.Q.;Wang,D.J.;Xin,B.F.;Li,S.D.;Sun,J.Z.Appl.Catal.B:Environ.2006,62,282.doi:10.1016/j.apcatb.2005.08.012
(31)Zhang,X.;Zhang,L.Z.;Xie,T.F.;Wang,D.J.J.Phys.Chem.C 2009,113,7371.doi:10.1021/jp900812d
(32)Kronik,L.;Shapira,Y.Surf.Sci.Rep.1999,37,1.doi:10.1016/S0167-5729(99)00002-3
(33)Lu,Y.C.;Wang,L.L.;Wang,D.J.;Xie,T.F.;Chen,L.P.;Lin,Y.H.Mater.Chem.Phys.2011,129,281.doi:10.1016/j.matchemphys.2011.04.004
猜你喜欢
环境卫生工程(2021年5期)2021-11-20
现代畜牧科技(2021年8期)2021-10-13
发光学报(2021年7期)2021-07-23
中成药(2017年7期)2017-11-22
魅力中国(2017年11期)2017-09-18
中国卫生(2016年7期)2016-11-13
当代化工研究(2016年2期)2016-03-20
中国卫生(2015年10期)2015-11-10
中国水利(2015年4期)2015-02-28
科学中国人(2015年22期)2015-02-28
